مرض الثلاسيميا: رحلة طبية مفصلة من الأعراض المبكرة إلى أحدث بروتوكولات العلاج
هل تحمل جيناته دون أن تدري؟ كل ما تحتاج معرفته عن أنيميا البحر المتوسط وخيارات علاجها

مرض الثلاسيميا اضطراب وراثي مزمن يصيب سلاسل الغلوبين (Globin Chains) المكوّنة للهيموغلوبين، فيُنتج الجسم كريات دم حمراء هشة وقصيرة العمر. يُصنَّف ضمن أكثر أمراض الدم الوراثية شيوعاً عالمياً، ويحمل جيناته نحو 270 مليون شخص حول العالم وفق تقديرات منظمة الصحة العالمية.
د. فارس أحمد الرشيدي — طبيب مختص في الأمراض الوراثية والجينوم
د. سوزان عبد الحميد السعدي — استشارية أمراض الدم وزرع الخلايا الجذعية
⚡ أهم ما ستخرج به من هذا المقال
🔬 الحقيقة العلمية الجوهرية
- الثلاسيميا ليست نقص حديد — العطل في مصنع بروتين الدم نفسه، والحديد لا يعالجها بل يضرّ.
- حامل السمة لا يحتاج علاجاً، لكنه يحمل مسؤولية وراثية عند اختيار شريك الحياة.
- تراكم الحديد — وليس فقر الدم — هو السبب الأول للمضاعفات الخطيرة على المدى الطويل.
✅ خطوات عمل فورية
- أجرِ فحص الهيموغلوبين الكهربائي قبل الزواج — ليس CBC العادي وحده.
- إذا كنت مريضاً: الفيريتين كل 3 أشهر + رنين قلب وكبد T2* سنوياً إلزامي.
- لا تتناول مكملات الحديد إطلاقاً دون تأكيد مخبري من طبيبك.
🚨 علامات تستدعي الطوارئ فوراً
- حمى فوق 38.5 درجة لمريض مستأصل الطحال — خطر تسمّم دم يُهدد الحياة.
- ضيق تنفس مفاجئ أو بول داكن اللون — توجّه للطوارئ فوراً.
🌟 بصيص الأمل العلمي
- العلاجات الجينية (Casgevy® وZynteglo®) حصلت على موافقة FDA 2023 وتُبشّر بشفاء حقيقي.
- الالتزام بنقل الدم والاستخلاب رفع العمر المتوقع من أقل من 20 عاماً إلى أكثر من 50 عاماً.
هل أخبرك أحدهم يوماً أن طفلك يعاني من “فقر دم لا يستجيب للحديد”؟ أو ربما اكتشفت في تحليل ما قبل الزواج أنك حامل لسمة الثلاسيميا ولم تفهم ماذا يعني ذلك لمستقبل أسرتك. الارتباك طبيعي، والخوف مشروع. لكنّ الفهم الدقيق لهذا المرض يحوّل القلق إلى خطة عمل واضحة. ستجد في هذا المقال إجابات عملية تساعدك على التمييز بين أنواعه، والتعرف على أعراضه مبكراً، واتخاذ قرارات صحية مبنية على علم لا على تخمين.
تخيّل أن أحمد — شاب سعودي في الثالثة والعشرين — ذهب مع خطيبته لإجراء فحص الثلاسيميا قبل الزواج في أحد مراكز الفحص المعتمدة بالرياض. جاءت النتيجة: كلاهما حامل لسمة بيتا ثلاسيميا الصغرى (Beta Thalassemia Minor). لم يشعر أيّ منهما بأعراض قط، لكنّ الطبيب أوضح لهما أن احتمال إنجاب طفل مصاب بالثلاسيميا الكبرى يبلغ 25% في كل حمل. أمام أحمد الآن خياران: إما استشارة وراثية متخصصة لفهم الخيارات الإنجابية المتاحة، وإما تجاهل النتيجة والمضي قدماً دون وعي بالمخاطر. الخلاصة العملية: لا تتجاهل نتيجة تحليل الثلاسيميا مهما بدت بسيطة — فالمعلومة التي تملكها اليوم قد تحمي أطفالك غداً.
ما هو مرض الثلاسيميا حقاً؟
التعريف العلمي المبسّط: خلل في إنتاج الهيموغلوبين
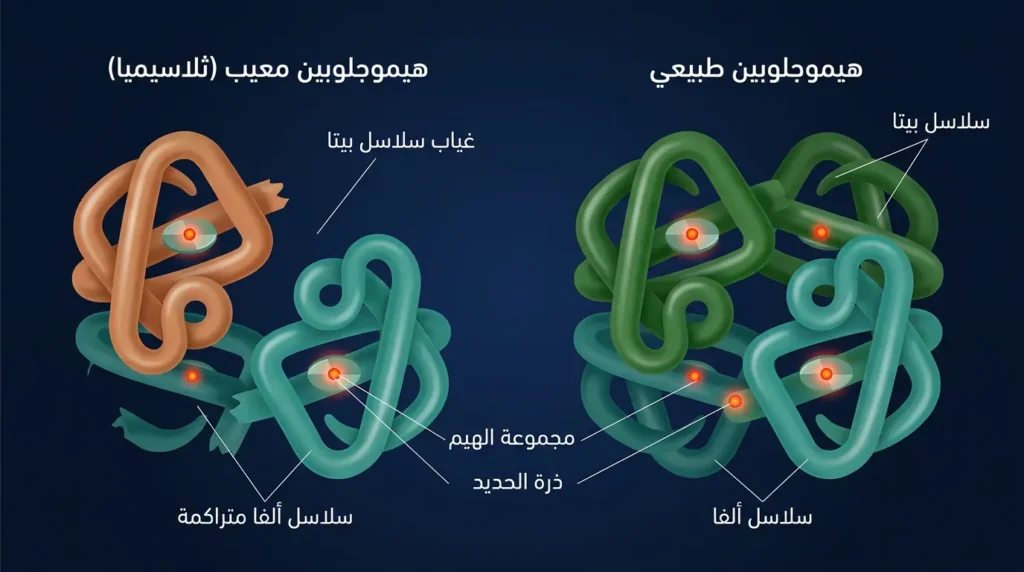
التعريف العلمي المبسّط: خلل في إنتاج الهيموغلوبين
لنبدأ من الأساس. الهيموغلوبين (Hemoglobin) هو البروتين الذي يمنح دمك لونه الأحمر، ووظيفته الجوهرية حمل الأكسجين من رئتيك إلى كل خلية في جسمك ثم إعادة ثاني أكسيد الكربون إلى الرئتين للتخلّص منه. تخيّل الهيموغلوبين وكأنه شاحنة نقل صغيرة تدور في شوارع جسدك؛ إذا كانت الشاحنة مصنوعة من مواد رديئة، فستتحطّم في منتصف الطريق ولن يصل الأكسجين إلى وجهته.
يتكوّن جزيء الهيموغلوبين الطبيعي من أربع سلاسل بروتينية: سلسلتَيْ ألفا غلوبين (Alpha Globin) وسلسلتَيْ بيتا غلوبين (Beta Globin). في مرض الثلاسيميا يحدث خلل في الجينات المسؤولة عن تصنيع إحدى هاتين السلسلتين، فيقلّ إنتاجها أو ينعدم تماماً. النتيجة؟ كريات دم حمراء هشّة، صغيرة الحجم، قصيرة العمر، وغير قادرة على أداء مهمتها. هذا هو جوهر فقر الدم الوراثي الذي نتحدث عنه.
والفارق الجوهري بين فقر الدم العادي والثلاسيميا أن الأوّل غالباً ناتج عن نقص الحديد أو الفيتامينات ويمكن علاجه بالمكملات، بينما الثاني عيب في “مصنع البروتين” نفسه داخل نخاع العظم، ولا يمكن إصلاحه بحبة حديد. لذلك إن وصف لك طبيب حديداً لعلاج فقر دم اتّضح لاحقاً أنه ثلاسيميا — فالحديد لن يفيد بل قد يضر.
لماذا يُعرف أيضاً باسم “أنيميا البحر المتوسط”؟
الاسم ليس اعتباطياً. اكتُشف المرض لأوّل مرّة بين عائلات من أصول يونانية وإيطالية في عشرينيات القرن العشرين، ومصطلح “Thalassa” يعني “البحر” باليونانية. انتشرت الطفرات الجينية المسبّبة للمرض بنسب مرتفعة في حوض البحر الأبيض المتوسط — إيطاليا واليونان وتركيا ولبنان وسوريا ومصر — وكذلك في شبه الجزيرة العربية وجنوب شرق آسيا وأجزاء من إفريقيا. في المملكة العربية السعودية تحديداً، تُشير بيانات وزارة الصحة إلى أن نسبة حاملي سمة الثلاسيميا تتراوح بين 3% و5% من السكان، وترتفع في المنطقة الشرقية وبعض مناطق الجنوب.
الجدير بالذكر أن سبب انتشار هذه الطفرات في هذه المناطق تحديداً يرتبط بارتباط وثيق بمرض الملاريا؛ إذ إنّ حمل سمة الثلاسيميا (دون الإصابة الكاملة) يمنح صاحبه مقاومة جزئية لطفيل الملاريا (Plasmodium)، مما جعل حاملي السمة أكثر قدرة على البقاء في المناطق الموبوءة بالملاريا عبر الأجيال.
حقيقة طبية: أنيميا البحر المتوسط ليست مقتصرة على سكان المتوسط فحسب. تُسجَّل حالات كثيفة في الهند والصين وباكستان وتايلاند، مما يجعل مرض الثلاسيميا مشكلة صحية عالمية لا إقليمية.
كيف ينتقل المرض؟ لغز الوراثة والجينات
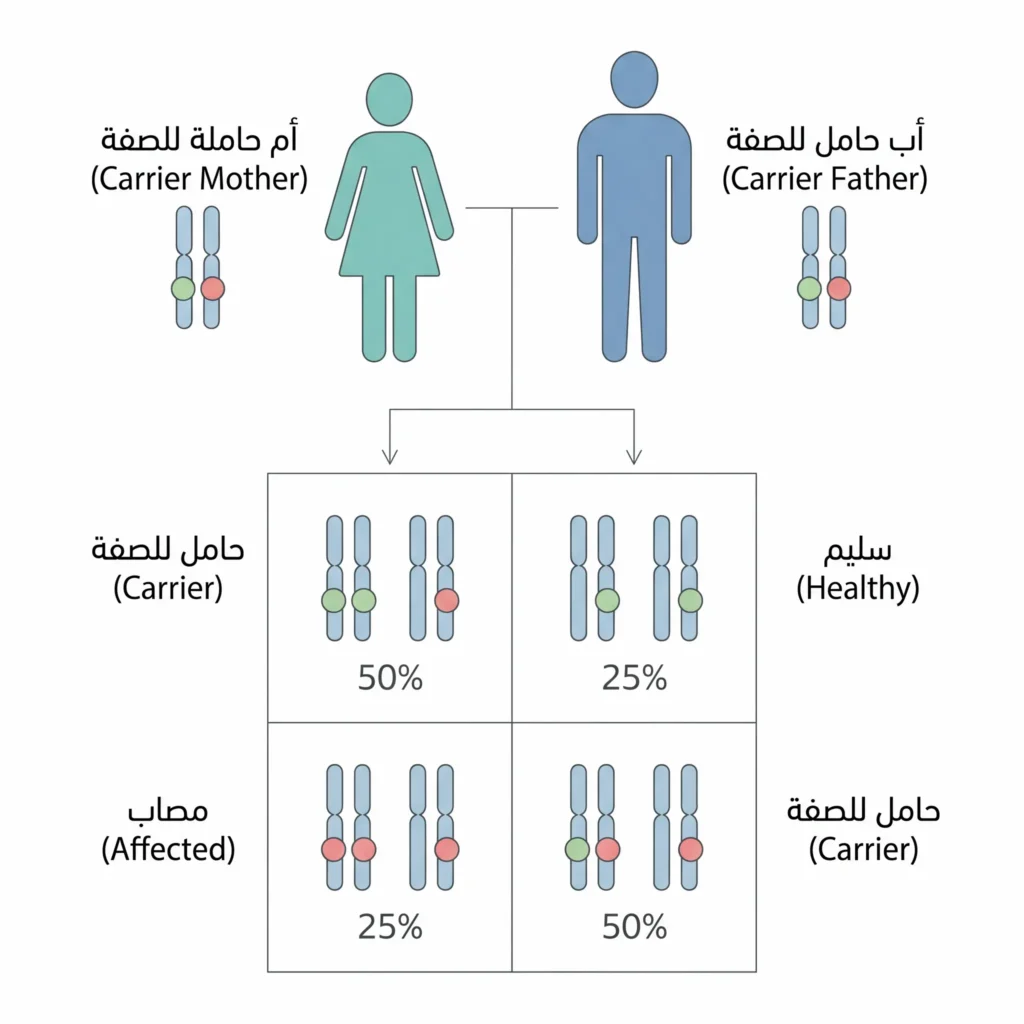
ما دور الجينات في تصنيع الدم؟
كل خلية في جسمك تحمل نسختين من كل جين: واحدة ورثتها من أمك والأخرى من أبيك. الجينات المسؤولة عن تصنيع سلاسل ألفا غلوبين تقع على الكروموسوم 16 (أربع نسخ)، بينما جينات بيتا غلوبين تقع على الكروموسوم 11 (نسختان). عندما يحدث خلل — حذف أو طفرة نقطية — في واحد أو أكثر من هذه الجينات، ينخفض إنتاج السلسلة المقابلة أو يتوقف كلياً. الخلل هنا ليس مكتسباً ولا معدياً؛ هو مكتوب في شفرتك الوراثية منذ لحظة الإخصاب.
كيف ينتقل المرض من الآباء إلى الأبناء؟
ينتقل مرض الثلاسيميا بنمط وراثة جسمية متنحية (Autosomal Recessive). ماذا يعني ذلك ببساطة؟ يعني أنك تحتاج إلى نسختين معطوبتين من الجين — واحدة من كل والد — لكي تظهر عليك أعراض المرض الكاملة. إذا ورثت نسخة معطوبة واحدة فقط، فستكون حامل الثلاسيميا (Carrier)؛ تحمل الجين لكنك غالباً لا تشعر بشيء يُذكر.
فلنُبسّط الأمر بمثال عملي: إذا كان الأب حاملاً للسمة والأم حاملة للسمة أيضاً، ففي كل حمل تكون الاحتمالات:
- 25% طفل سليم تماماً.
- 50% طفل حامل للسمة (مثل والديه).
- 25% طفل مصاب بالثلاسيميا الكبرى.
هذه النسب لا تعني أن من بين كل أربعة أطفال سيكون واحد مصاباً حتماً؛ بل تعني أن كل حمل على حدة يحمل هذا الاحتمال، تماماً كرمي قطعة نقدية — كل رمية مستقلة عن سابقتها.
ما الفرق الجوهري بين “حامل المرض” و”المصاب بالمرض”؟
هنا نصل إلى نقطة محورية كثيراً ما تختلط على الناس. حامل الثلاسيميا (يُسمّى أيضاً: لديه سمة الثلاسيميا أو Thalassemia Trait) يملك نسخة جينية سليمة وأخرى معطوبة. جسمه يُنتج هيموغلوبيناً كافياً — وإن كان أقل قليلاً من الطبيعي — ويعيش حياة طبيعية تماماً دون أعراض أو مع أعراض خفيفة جداً كانخفاض طفيف في الهيموغلوبين. على النقيض من ذلك، المصاب بالمرض (خاصة الثلاسيميا الكبرى) يملك نسختين معطوبتين، فيكون إنتاج الهيموغلوبين ناقصاً على نحو شديد ويحتاج إلى نقل دم دوري مدى الحياة.
النصيحة العملية: إذا اكتشفت أنك حامل للسمة، فلا داعي للقلق على صحتك الشخصية. لكنّ المسؤولية تبدأ عندما تختار شريك حياتك؛ لأن زواج حاملين اثنين هو ما يُنتج أطفالاً مصابين.
| وجه المقارنة | حامل السمة (الصغرى) | المصاب بالكبرى |
|---|---|---|
| الجينات المتأثرة | نسخة واحدة معطوبة فقط | نسختان معطوبتان |
| الأعراض السريرية | لا أعراض أو أعراض بالغة الخفّة | شحوب، خمول، تضخم أعضاء، تشوهات عظام |
| مستوى الهيموغلوبين | طبيعي أو منخفض قليلاً (10-12 غ/دل) | منخفض جداً (< 7 غ/دل بدون علاج) |
| الحاجة لنقل الدم | لا | نعم، كل 2-4 أسابيع مدى الحياة |
| الحاجة لأدوية الاستخلاب | لا | نعم، ضرورة حياتية |
| الخطر الوراثي | 25% خطر إنجاب طفل مصاب إن تزوج من حامل | 100% من أبنائه حاملون على الأقل |
| جودة الحياة | طبيعية تماماً | تعتمد على الالتزام العلاجي |
| خطأ شائع يجب تجنبه | وصف مكملات الحديد بالخطأ | إعطاء حديد — خطر جسيم على حياته |
| إمكانية الشفاء | لا يحتاج علاجاً | زراعة نخاع العظم أو العلاج الجيني |
اقرأ أيضاً:
ما الذي يجعل الثلاسيميا الصغرى مختلفة جذرياً عن الكبرى؟
هذا هو السؤال الذي يدخل كثير من القراء إلى المقال بحثاً عن إجابته، وهو يستحق تفصيلاً وافياً. الفرق بين الثلاسيميا الكبرى والصغرى ليس مجرد فرق في الدرجة، بل هو فرق في نمط الحياة بأكمله.
الثلاسيميا الصغرى (Minor) لا تحتاج عادةً إلى أي علاج. قد يُظهر تحليل الدم انخفاضاً طفيفاً في حجم كريات الدم (MCV أقل من 80 فيمتولتر) وربما انخفاضاً بسيطاً في الهيموغلوبين، لكن صاحبها يمارس حياته ورياضته ويعمل ويسافر دون عوائق تُذكر. الخطأ الشائع هنا أن بعض الأطباء — مع الأسف — يصفون الحديد لهؤلاء الأشخاص ظناً أنهم يعانون فقر دم ناتج عن نقص الحديد، وهذا قد يؤدي إلى تراكم حديد غير ضروري.
أما الثلاسيميا الكبرى (Major) — وتُسمّى أيضاً أنيميا كولي (Cooley’s Anemia) — فهي قصة مختلفة تماماً. الطفل يولد طبيعياً في الأشهر الأولى لأن الهيموغلوبين الجنيني (Hemoglobin F) يؤدي المهمة مؤقتاً، لكن بعد الشهر السادس تقريباً — حين يُفترض أن يتحوّل الجسم لإنتاج الهيموغلوبين البالغ (Hemoglobin A) — تبدأ الأعراض: شحوب شديد، خمول، رفض الرضاعة، تأخر نمو. وبدون نقل دم منتظم كل أسبوعين إلى أربعة أسابيع، يهبط الهيموغلوبين إلى مستويات خطيرة تهدد الحياة.
الثلاسيميا الوسطى (Intermedia) تقع بين الطرفين: أعراض أوضح من الصغرى لكنها لا تستدعي نقل دم دورياً في كل الحالات، وإنما قد يحتاج المريض لنقل دم متقطع في أوقات الضغط كالعدوى أو الجراحة أو الحمل. وهناك تفاوت واسع ضمن هذه الفئة يعتمد على نوع الطفرة الجينية تحديداً.
معلومة سريعة: ليست كل طفرات بيتا غلوبين متساوية في الشدة. بعض الطفرات تُوقف إنتاج السلسلة كلياً (β⁰)، وبعضها يقلّل الإنتاج فقط (β⁺). هذا الفرق الجزيئي الدقيق هو ما يحدد إن كنت ستحتاج نقل دم شهرياً أم ستعيش بأعراض خفيفة فقط.
التصنيف الطبي الدقيق لأنواع الثلاسيميا
ألفا ثلاسيميا (Alpha Thalassemia) وتدرجاتها
تنتج ألفا ثلاسيميا عن حذف (Deletion) واحد أو أكثر من الجينات الأربعة المسؤولة عن سلسلة ألفا غلوبين على الكروموسوم 16. كلما زاد عدد الجينات المحذوفة، زادت شدة المرض:
الحامل الصامت (Silent Carrier): حذف جين واحد فقط. لا أعراض البتّة، ولا يُكتشف عادةً إلا بالفحص الجيني. تحليل الدم الروتيني يبدو طبيعياً تقريباً.
سمة ألفا ثلاسيميا (Alpha Thalassemia Trait): حذف جينين. قد يظهر انخفاض طفيف في حجم كريات الدم وفي نسبة الهيموغلوبين، لكن الشخص يعيش حياة طبيعية. وهذه هي الحالة التي تُكتشف أحياناً بالصدفة عند إجراء تحليل CBC لأي سبب آخر.
مرض هيموغلوبين H (Hemoglobin H Disease): حذف ثلاثة جينات. هنا يبدأ المريض بمعاناة حقيقية: فقر دم متوسط إلى شديد، تضخم في الطحال، واحتمال الحاجة لنقل دم أحياناً. يتشكّل هيموغلوبين غير طبيعي يُسمّى هيموغلوبين H (مكوّن من أربع سلاسل بيتا غلوبين بدلاً من سلسلتي ألفا وسلسلتي بيتا)، وهو غير فعّال في نقل الأكسجين.
الاستسقاء الجنيني (Hydrops Fetalis): حذف الجينات الأربعة كلها. هذا أخطر أنواع الثلاسيميا على الإطلاق، ويكون غالباً غير متوافق مع الحياة. الجنين يعاني من فقر دم شديد جداً واستسقاء معمّم (تجمّع سوائل في أنحاء الجسم) ويتوفّى عادةً في الرحم أو بعد الولادة بوقت قصير. هذا النوع يتطلّب تشخيصاً مبكراً في أثناء الحمل عبر الفحوصات الجينية.
بيتا ثلاسيميا (Beta Thalassemia) وتدرجاتها
تنتج بيتا ثلاسيميا عن طفرات نقطية (Point Mutations) — لا حذف كامل عادةً — في جينات بيتا غلوبين على الكروموسوم 11. تُصنّف إلى:
الثلاسيميا الصغرى (Beta Thalassemia Minor / Trait): طفرة في نسخة واحدة فقط من الجين. فقر دم خفيف، كريات دم صغيرة، لا أعراض سريرية ملموسة عادةً. لا يحتاج صاحبها لعلاج بل فقط لمتابعة ووعي وراثي.
الثلاسيميا الوسطى (Beta Thalassemia Intermedia): طفرات في النسختين لكنها أقل شدة، أو وجود عوامل وراثية مساعدة (مثل ارتفاع الهيموغلوبين الجنيني F) تُخفّف حدة المرض. الأعراض متفاوتة: قد يحتاج المريض لنقل دم عَرَضي وليس دورياً.
الثلاسيميا الكبرى / أنيميا كولي (Beta Thalassemia Major / Cooley’s Anemia): طفرات شديدة في النسختين تُعطّل إنتاج بيتا غلوبين بالكامل تقريباً. تظهر الأعراض في السنة الأولى من العمر. نقل الدم الدوري ضرورة حياتية، ومعه يأتي تحدّي تراكم الحديد الذي يستلزم علاجاً مستمراً بأدوية الاستخلاب. دون علاج، لا يتجاوز العمر المتوقع السنوات القليلة الأولى.
| النوع | الجينات المتأثرة | شدة الأعراض | الأعراض الرئيسية | الحاجة لنقل الدم | الخيار العلاجي |
|---|---|---|---|---|---|
| الحامل الصامت (ألفا) | 1 جين محذوف | لا أعراض | لا شيء | لا | وعي وراثي فقط |
| سمة ألفا ثلاسيميا | 2 جين محذوف | خفيفة جداً | انخفاض طفيف في MCV | لا | متابعة دورية |
| مرض هيموغلوبين H | 3 جينات محذوفة | متوسطة | فقر دم، تضخم طحال | أحياناً | حمض فوليك، متابعة |
| استسقاء الجنين | 4 جينات محذوفة | مميت | فقر دم حاد، وفاة جنينية | — | تشخيص ما قبل الولادة |
| بيتا صغرى (سمة) | طفرة في نسخة واحدة | لا أعراض عملياً | ارتفاع HbA2 فوق 3.5% | لا | وعي وراثي فقط |
| بيتا وسطى | طفرتان أقل شدة | متوسطة | فقر دم، تضخم طحال متغير | متقطع | استخلاب، فوليك، متابعة |
| بيتا كبرى (أنيميا كولي) | طفرتان شديدتان | شديدة جداً | فقر دم حاد، تشوهات عظام، تضخم أعضاء | كل 2-4 أسابيع | نقل دم + استخلاب + زراعة نخاع |
رقم لافت: تُقدّر منظمة الصحة العالمية أن نحو 60,000 طفل يُولدون سنوياً مصابين بأشكال شديدة من بيتا ثلاسيميا، ويتركّز معظمهم في جنوب وجنوب شرق آسيا ومنطقة الشرق الأوسط.
المختبر الفسيولوجي — للمهتمين بالتفاصيل العلمية الدقيقة
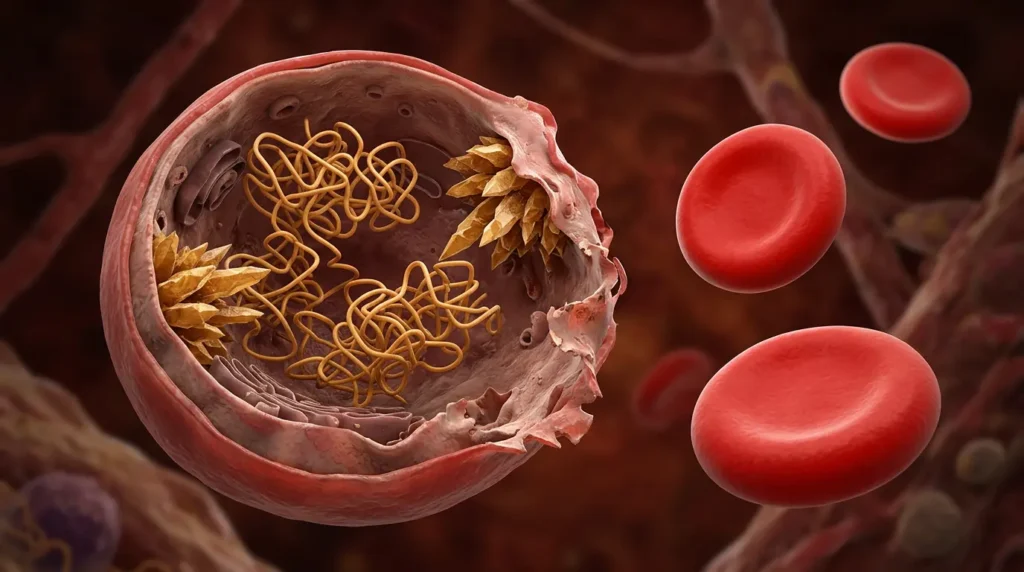
لنغُص أعمق في ما يحدث داخل نخاع العظم والدورة الدموية عند مريض الثلاسيميا. في الوضع الطبيعي، تُنتج الخلايا السلفية الحمراء (Erythroid Precursors) في نخاع العظم سلاسل ألفا وبيتا غلوبين بنسب متساوية ومتزامنة. هذا التوازن حاسم؛ لأن أي سلسلة غلوبين حرّة — غير مرتبطة بنظيرتها — تصبح غير مستقرة وتترسّب داخل الخلية.
في بيتا ثلاسيميا الكبرى مثلاً، يتوقف إنتاج سلاسل بيتا غلوبين أو يتراجع بشدة. النتيجة: تتراكم سلاسل ألفا غلوبين الزائدة داخل الخلية الحمراء النامية. هذه السلاسل الحرّة شديدة السمّية الخلوية؛ إذ تتأكسد وتشكّل ما يُعرف بالشوائب التأكسدية (Oxidative Inclusions) التي تُتلف غشاء الخلية الحمراء من الداخل. تخيّل قطعاً معدنية صدئة تدور داخل بالون مطاطي — ستثقب البالون حتماً.
هذا التلف يُفضي إلى ظاهرتين مترابطتين. الأولى: تكوّن الدم غير الفعّال (Ineffective Erythropoiesis)، أي أن نسبة كبيرة من كريات الدم الحمراء تموت داخل نخاع العظم قبل أن تنضج وتخرج إلى الدورة الدموية. الثانية: الكريات التي تنجو وتخرج إلى الدم تكون هشّة ومشوّهة، فيلتقطها الطحال ويدمّرها بسرعة — وهو ما يُسمّى انحلال الدم المحيطي (Peripheral Hemolysis). الطحال يعمل هنا وكأنه مصفاة تلتقط الكريات المعيبة، ومع مرور الوقت يتضخّم بسبب العمل الإضافي المفروض عليه.
كما أن نخاع العظم يحاول تعويض النقص بتوسيع نشاطه بشدة — وهو ما يُسمّى فرط تكوّن الدم التعويضي (Compensatory Erythroid Hyperplasia). هذا التوسّع يُضعف بنية العظام من الداخل لأنه يحتلّ فراغات أكبر داخل التجويف النخاعي، مما يفسّر هشاشة العظام وتشوّهات الوجه والجمجمة المميزة (مثل بروز الجبهة وعظام الوجنتين) التي تُشاهَد في الحالات الشديدة غير المعالجة.
على الصعيد الجزيئي، يلعب مسار الإريثروبويتين (Erythropoietin – EPO) دوراً مركزياً. الكلية تستشعر نقص الأكسجين فتُفرز كميات ضخمة من هرمون EPO لتحفيز نخاع العظم على إنتاج المزيد من كريات الدم. لكنّ المشكلة أن هذا “الإنتاج الزائد” معيب في أساسه — فيدخل الجسم في حلقة مفرغة: نقص أكسجين → إنتاج مفرط → كريات معيبة → تدمير مبكر → نقص أكسجين مجدداً.
من ناحية أخرى، يزيد الامتصاص المعوي للحديد بفعل تثبيط هرمون الهيبسيدين (Hepcidin) — وهو الهرمون الكبدي المسؤول عن تنظيم دخول الحديد إلى الدم. في الثلاسيميا، يُنتج نخاع العظم المُجهَد بروتينات مثل الإريثروفيرون (Erythroferrone – ERFE) تُثبّط الهيبسيدين، فيُفتح الباب أمام امتصاص حديد زائد حتى بدون نقل دم. هذا الحديد الزائد يترسّب في القلب والكبد والغدد الصماء مسبباً أضراراً تراكمية خطيرة.
ومضة علمية: أثبتت دراسة منشورة في مجلة Blood عام 2019 أن مستوى الإريثروفيرون (ERFE) يرتبط طردياً بشدة فقر الدم في مرضى بيتا ثلاسيميا الوسطى، وأن استهداف هذا البروتين علاجياً قد يُعيد تنظيم امتصاص الحديد.
اقرأ أيضاً:
ما الأعراض والعلامات التحذيرية التي تدلّ على الثلاسيميا؟
الأعراض الصامتة والمبكرة عند الرضع
كما ذكرنا، الرضيع المصاب بالثلاسيميا الكبرى يبدو طبيعياً عند الولادة. السبب أن الهيموغلوبين الجنيني (HbF) لا يحتاج سلاسل بيتا غلوبين (يتكوّن من سلسلتي ألفا وسلسلتي غاما). لكن بين الشهر الثالث والسادس من العمر — حين يبدأ الجسم بالتحوّل لإنتاج الهيموغلوبين البالغ (HbA) — تظهر أولى العلامات:
شحوب تدريجي في لون البشرة والأغشية المخاطية. الأم تلاحظ أن طفلها “أصفر” أو “باهت” دون سبب واضح. خمول وقلة نشاط مقارنة بأقرانه. صعوبة في الرضاعة أو فقدان الشهية. بكاء مستمر وانزعاج قد يُفسَّر خطأً على أنه مغص.
هذه الأعراض ليست حصرية للثلاسيميا بالطبع، لكنها تستدعي إجراء تحليل دم بسيط (CBC) يكشف فقر الدم وصغر حجم الكريات. إذا كنتِ أمّاً ولاحظتِ أن طفلك شاحب على نحو غير اعتيادي ولم يتحسّن بعد أسبوعين من المتابعة — اطلبي من طبيب الأطفال إجراء فحص الهيموغلوبين الكهربائي (Hemoglobin Electrophoresis) وليس فقط CBC.
اقرأ أيضاً:
- اليرقان الوليدي (صفار المواليد): الأسباب، درجات الخطورة، وخطوات العلاج الطبية
- انحلال الدم الوليدي: الأسباب، الأعراض، وخطوات العلاج والوقاية
الأعراض المتقدمة والظاهرة
عندما يتأخر التشخيص أو يكون العلاج غير كافٍ، تتطور أعراض الثلاسيميا لتشمل علامات أوضح:
تضخّم الطحال والكبد (Hepatosplenomegaly): يتضخّم الطحال لأنه يعمل فوق طاقته في تدمير الكريات المعيبة، ويتضخّم الكبد لأنه يتحمّل عبئاً إضافياً في تصنيع الدم خارج نخاع العظم (تكوّن الدم خارج النخاع — Extramedullary Hematopoiesis).
تشوّهات العظام: بروز عظام الجبهة والوجنتين، وتوسّع التجويف النخاعي في عظام الجمجمة (ما يُعرف إشعاعياً بمظهر “فروة الشعر القائمة” أو Hair-on-End Appearance في صور الأشعة). هذه التغييرات تمنح الوجه ملامح مميزة يُطلق عليها أحياناً “الوجه الثلاسيمي” (Thalassemic Facies).
تأخّر النمو والبلوغ: فقر الدم المزمن وسوء إيصال الأكسجين وتراكم الحديد في الغدد الصمّاء (كالغدة النخامية والغدة الدرقية والمبيضين والخصيتين) يؤدّي إلى قِصَر القامة وتأخّر ظهور علامات البلوغ.
لون البشرة الداكن أو المائل للأصفر: بسبب انحلال الدم المزمن وارتفاع البيليروبين غير المباشر (Indirect Bilirubin).
حصوات المرارة: الانحلال المزمن يُنتج كميات كبيرة من البيليروبين التي قد تترسّب في المرارة مكوّنةً حصوات صبغية (Pigment Gallstones) حتى في سن الطفولة.
نقطة تستحق الانتباه: ليست كل أعراض الثلاسيميا ناتجة عن المرض ذاته؛ كثير منها ناتج عن تراكم الحديد الذي يأتي كأثر جانبي لعمليات نقل الدم المتكررة. لذلك العلاج لا يتوقف عند نقل الدم، بل يشمل حتماً التحكّم في مخزون الحديد.
اقرأ أيضاً:
متى يجب عليك التوجه للطوارئ أو زيارة الطبيب فوراً؟
ليست كل أعراض مرض الثلاسيميا تستدعي غرفة الطوارئ، لكنّ بعض العلامات تمثّل إنذاراً أحمر لا يحتمل التأجيل. إذا كان طفلك أو أحد أفراد عائلتك مصاباً بالثلاسيميا، فانتبه لما يلي:
حمّى أعلى من 38.5 درجة مئوية: خاصة إذا كان المريض قد خضع لاستئصال الطحال (Splenectomy)؛ لأنه يكون معرّضاً بشدة لعدوى بكتيرية خاطفة قد تتطور إلى تسمّم دموي (Sepsis) خلال ساعات.
ضيق تنفّس مفاجئ أو خفقان سريع غير معتاد: قد يشير إلى هبوط حاد في الهيموغلوبين أو مشكلة قلبية مرتبطة بتراكم الحديد في عضلة القلب.
ألم شديد في البطن مع انتفاخ: قد يدلّ على احتشاء الطحال (Splenic Infarction) أو تمزّقه، خاصة في الحالات المصحوبة بتضخّم شديد.
إغماء أو دوخة شديدة لا تتحسّن بالراحة: علامة على فقر دم حاد يحتاج نقل دم طارئ.
تغيّر لون البول إلى بني داكن أو أحمر: قد يشير إلى أزمة انحلالية حادة (Hemolytic Crisis).
النصيحة المباشرة: إذا لاحظت أياً من هذه العلامات — لا تنتظر حتى الصباح أو حتى موعد العيادة القادم. توجّه إلى أقرب طوارئ فوراً واحمل معك ملفّ المريض الطبي وآخر تحاليل دم.
اقرأ أيضاً:
كيف يشخّص الأطباء مرض الثلاسيميا؟
الفحوصات الروتينية: صورة الدم الكاملة ومسحة الدم
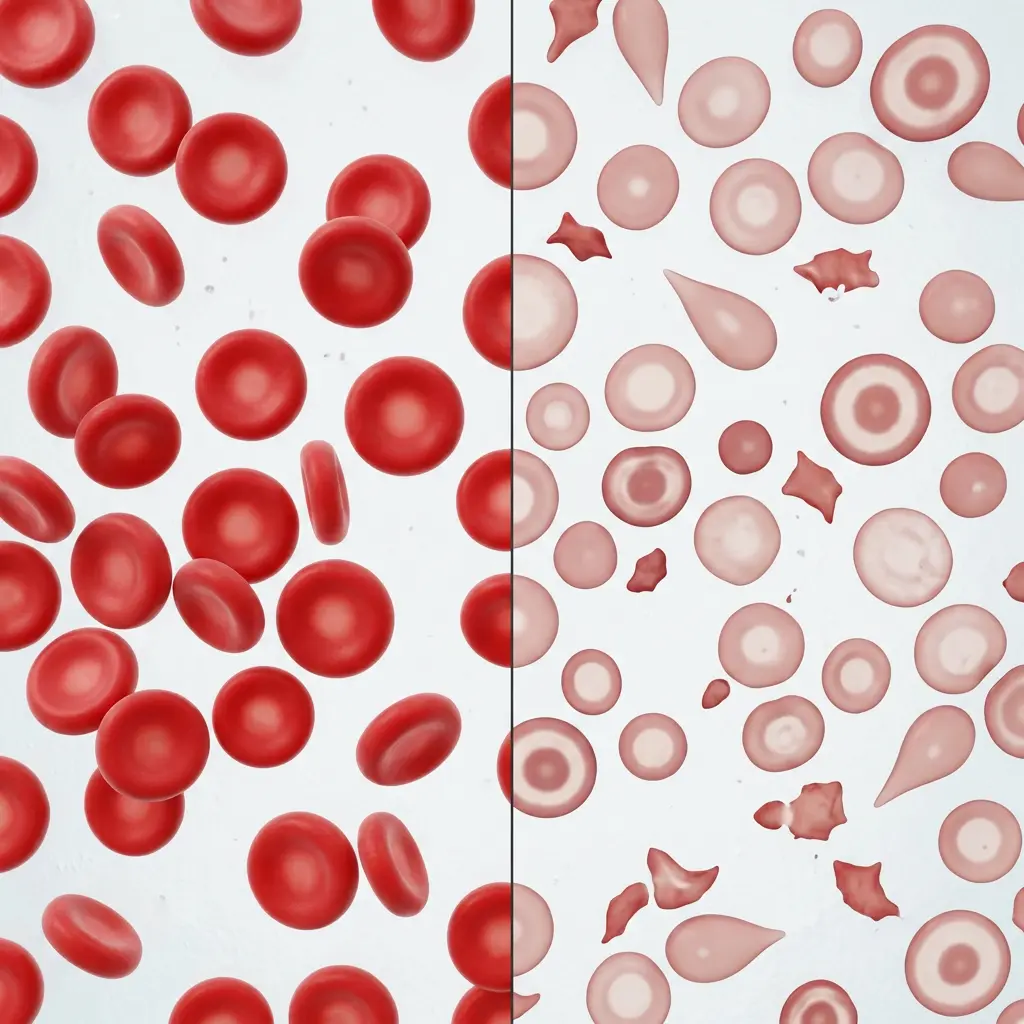
أوّل خطوة تشخيصية هي تحليل صورة الدم الكاملة (Complete Blood Count — CBC). في الثلاسيميا ستلاحظ النتائج التالية عادةً:
انخفاض متوسط حجم الكرية الحمراء (MCV) — عادةً أقل من 75-80 فيمتولتر. انخفاض متوسط محتوى الهيموغلوبين في الكرية (MCH). ارتفاع عدد كريات الدم الحمراء أحياناً رغم انخفاض الهيموغلوبين (وهذا فارق تشخيصي عن فقر الدم بنقص الحديد الذي ينخفض فيه العدد عادةً).
مسحة الدم (Blood Smear) تحت المجهر تكشف كريات حمراء صغيرة (Microcytic)، شاحبة (Hypochromic)، وقد تظهر “خلايا هدفية” (Target Cells) وهي كريات ذات شكل مميز يُشبه الهدف، بالإضافة إلى كريات مشوّهة ودموع الكريات (Teardrop Cells).
الفحوصات المتقدمة: الفصل الكهربائي للهيموغلوبين
تحليل الثلاسيميا الأكثر تحديداً هو الفصل الكهربائي للهيموغلوبين (Hemoglobin Electrophoresis) أو تحليل HPLC (High-Performance Liquid Chromatography). هذا الفحص يفصل أنواع الهيموغلوبين الموجودة في دمك ويحدد نسبة كل نوع. في بيتا ثلاسيميا الصغرى مثلاً، ترتفع نسبة الهيموغلوبين A2 (HbA2) فوق 3.5%، وقد ترتفع نسبة الهيموغلوبين الجنيني (HbF) قليلاً.
هذا الفحص متوفر في معظم المستشفيات الكبرى والمختبرات المعتمدة في السعودية، وهو الفحص المطلوب ضمن برنامج فحص ما قبل الزواج الذي أقرّته وزارة الصحة السعودية منذ عام 2004.
الفحص الجيني وفحوصات ما قبل الولادة
في حالات محددة — كأن يكون كلا الزوجين حاملين للسمة — يُوصى بالفحص الجيني التفصيلي (DNA Analysis / Molecular Genetic Testing) لتحديد نوع الطفرة بالضبط. هذا الفحص مهم لأنه يُنبئ بشدة المرض المتوقعة عند الطفل ويساعد في التخطيط العلاجي المبكر.
كما يمكن إجراء فحوصات ما قبل الولادة للجنين لتحديد إصابته:
- بزل السائل الأمينوسي (Amniocentesis): بين الأسبوعين 15-18 من الحمل.
- أخذ عينة من الزغابات المشيمية (Chorionic Villus Sampling — CVS): بين الأسبوعين 10-13.
- فحص الحمض النووي الحر في دم الأم (Non-Invasive Prenatal Testing — NIPT): وهو فحص أحدث وأقل خطورة، لكنه لا يزال محدود الدقة في تشخيص الثلاسيميا مقارنة بالطرق الباضعة.
يشير الدكتور فارس أحمد الرشيدي — طبيب مختص في الأمراض الوراثية والجينوم في موقع وصفة طبية — إلى أن “معرفة نوع الطفرة الجينية بدقة ليست ترفاً أكاديمياً، بل هي أداة إكلينيكية فعلية تساعد الطبيب على توقع مسار المرض واختيار البروتوكول العلاجي الأنسب لكل مريض على حدة، كما تُتيح للأسرة اتخاذ قرارات إنجابية واعية.”
خرافات شائعة وحقائق علمية عن الثلاسيميا
❌ الخرافة: مرض الثلاسيميا يمكن أن ينتقل بالعدوى أو بالتلامس مع المريض.
✅ الحقيقة: الثلاسيميا مرض وراثي جيني بالكامل، وينتقل فقط من الآباء إلى الأبناء عبر الجينات. لا علاقة له بالعدوى أو بالتلامس أو بالطعام أو بنمط الحياة. مصدر: منظمة الصحة العالمية — صحيفة وقائع فقر الدم المنجلي والثلاسيميا.
❌ الخرافة: إذا كانت نتيجة فحص الثلاسيميا “حامل”، فهذا يعني أنني مريض وأحتاج علاجاً.
✅ الحقيقة: حامل الثلاسيميا (صاحب السمة) ليس مريضاً. لديه نسخة جينية معطوبة وأخرى سليمة، وجسمه يعمل على نحو طبيعي تقريباً. لا يحتاج أي علاج، لكنه بحاجة لوعي وراثي عند اختيار شريك الحياة.
❌ الخرافة: مريض الثلاسيميا الكبرى يجب أن يتناول مكملات الحديد لأنه يعاني فقر دم.
✅ الحقيقة: هذا من أخطر المعتقدات. مريض الثلاسيميا الكبرى يعاني بالفعل من فقر دم، لكنّ المشكلة ليست نقص الحديد بل تدمير الكريات الحمراء. تناول الحديد في هذه الحالة يزيد تراكمه في الأعضاء ويُسرّع المضاعفات. العلاج الصحيح هو نقل الدم مع أدوية الاستخلاب لطرد الحديد الزائد.
❌ الخرافة: لا يمكن لمريض الثلاسيميا أن يعيش حياة طبيعية أو يتزوج أو ينجب.
✅ الحقيقة: مع التطور الطبي الحديث في علاجات نقل الدم والاستخلاب وزراعة نخاع العظم والعلاج الجيني، يعيش كثير من مرضى الثلاسيميا حياة طبيعية ومنتجة. في السعودية وحدها، يوجد مئات المرضى المتزوجين والعاملين والناشطين في مجتمعاتهم.
❌ الخرافة: فحص ما قبل الزواج “يكفي” لمنع المرض نهائياً.
✅ الحقيقة: الفحص لا يمنع المرض، بل يكشف الحاملين. القرار النهائي يبقى بيد الزوجين. إذا كان كلاهما حاملاً للسمة وقررا الزواج، فلا يزال احتمال إنجاب طفل مصاب قائماً (25% في كل حمل). الفحص أداة وعي لا أداة حظر.
ما مضاعفات الثلاسيميا الخطيرة وكيف نتجنبها؟
تراكم الحديد: العدو الخفي الأكبر
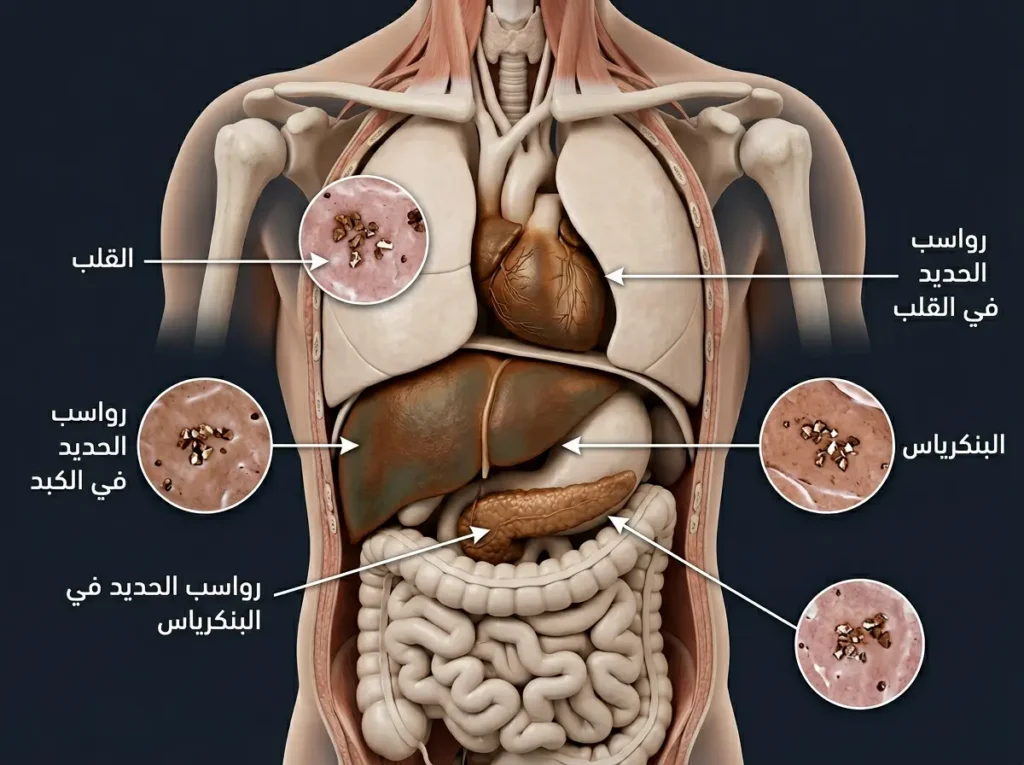
إذا كان هناك عنوان واحد يجب أن تتذكّره من هذا المقال بأكمله، فهو: تراكم الحديد. هذه هي المعركة الحقيقية لمريض الثلاسيميا الكبرى، وهي السبب الأول للمضاعفات المهددة للحياة على المدى الطويل.
كيف يتراكم الحديد؟ من مصدرين: أولاً من عمليات نقل الدم المتكررة (كل وحدة دم تحتوي على 200-250 ملغ من الحديد تقريباً)؛ وثانياً من زيادة الامتصاص المعوي بسبب تثبيط هرمون الهيبسيدين كما شرحنا في الفقرة الفسيولوجية. الجسم البشري لا يملك آلية فعّالة لإفراز الحديد الزائد، فيترسّب في الأعضاء ببطء وصمت.
أين يترسّب الحديد؟ في القلب (يسبب اعتلال عضلة القلب وفشل القلب واضطرابات النظم)؛ في الكبد (يسبب تليّفاً وتشمّعاً وأحياناً سرطان الكبد)؛ في الغدد الصمّاء (يسبب السكري وقصور الغدة الدرقية وتأخر البلوغ وهشاشة العظام). الحديد الزائد في القلب تحديداً كان تاريخياً السبب الأول لوفاة مرضى الثلاسيميا قبل توفّر أدوية الاستخلاب الحديثة.
مراقبة مخزون الحديد تتمّ عبر تحليل الفيريتين (Serum Ferritin) بشكل دوري كل 3 أشهر، وعبر تصوير الرنين المغناطيسي للقلب والكبد (MRI T2*) سنوياً لتقدير كمية الحديد المترسّبة في هذين العضوين بدقة.
ماذا تفعل الآن؟ إذا كان فيريتين المريض أعلى من 1000 نانوغرام/مل باستمرار، فهذا مؤشر واضح على حاجة لضبط جرعة أدوية الاستخلاب أو الالتزام الأفضل بها. تحدّث مع طبيب الدم المعالج فوراً.
تضخّم الطحال ومشاكل الكبد
الطحال المتضخّم لا يكتفي بتدمير الكريات المعيبة بل يبدأ بتدمير الكريات الطبيعية أيضاً والصفائح الدموية وكريات الدم البيضاء — وهو ما يُسمّى فرط نشاط الطحال (Hypersplenism). عندما يصل التضخّم لمرحلة تستلزم نقل دم بمعدل أعلى بكثير من المتوقع، قد يوصي الطبيب باستئصال الطحال. لكنّ هذا القرار ليس بسيطاً؛ لأن الطحال يلعب دوراً مناعياً مهمّاً، واستئصاله يجعل المريض عرضة لعدوى بكتيرية خطيرة بالمكوّرات الرئوية (Streptococcus pneumoniae) والنيسرية السحائية (Neisseria meningitidis) وغيرها.
هشاشة العظام
بين 40% و50% من مرضى الثلاسيميا البالغين يعانون من هشاشة عظام (Osteoporosis) أو انخفاض كثافة العظام (Osteopenia). الأسباب متشعّبة: فرط نشاط نخاع العظم الذي يُرقّق القشرة العظمية، ونقص هرمونات الغدد الصمّاء المتضررة بتراكم الحديد، ونقص فيتامين D والكالسيوم. ولذلك يُنصح بإجراء فحص كثافة العظام (DEXA Scan) بانتظام للبالغين المصابين.
مشاكل القلب المرتبطة بالمرض
القلب هو العضو الأكثر حساسية لتراكم الحديد. اعتلال عضلة القلب بالحديد (Iron Overload Cardiomyopathy) يتطور بصمت، وقد لا يشعر المريض بأعراض حتى يصل إلى مرحلة متقدمة. لحسن الحظ، إذا اكتُشف مبكراً عبر الرنين المغناطيسي (T2* أقل من 20 ميلي ثانية)، فإن العلاج المكثف بالاستخلاب يمكن أن يعكس الضرر ويُحسّن وظيفة القلب. هذه من الحقائق التي تبعث على الأمل: تلف القلب بالحديد قابل للعلاج إذا اكتُشف مبكراً.
صندوق الاقتباس الطبي:
وفق بيانات الاتحاد الدولي للثلاسيميا (Thalassaemia International Federation — TIF)، فإن متوسط العمر المتوقع لمرضى الثلاسيميا الكبرى الملتزمين بنقل الدم وأدوية الاستخلاب ارتفع من أقل من 20 عاماً في الثمانينيات إلى أكثر من 50 عاماً حالياً في الدول ذات الرعاية الطبية الجيدة. هذا التحسّن الدراماتيكي يعكس أثر الالتزام العلاجي.
اقرأ أيضاً:
- قصور القلب (ضعف عضلة القلب): الأسباب، العلامات التحذيرية، وخيارات العلاج المتقدمة
- عدم انتظام ضربات القلب: الأسباب، العلامات التحذيرية، وأحدث بروتوكولات العلاج
البروتوكولات العلاجية: من الإدارة اليومية إلى الأمل بالشفاء
عمليات نقل الدم الدورية: الضرورة والتحديات
⚠️ تحذير: الفقرة التالية تحتوي على معلومات دوائية وعلاجية مفصّلة. لا تبدأ أي علاج أو تُوقفه أو تُعدّل جرعته دون إشراف طبيبك المعالج.
نقل الدم المنتظم هو العمود الفقري لعلاج الثلاسيميا الكبرى. الهدف هو الحفاظ على مستوى هيموغلوبين ما قبل نقل الدم (Pre-transfusion Hemoglobin) بين 9.5 و10.5 غ/دل. هذا المستوى يكفي لمنع التوسّع النخاعي الزائد وتشوّهات العظام وتضخّم الطحال، ويسمح للطفل بالنمو والتطور بشكل طبيعي.
عادةً يحتاج المريض لنقل دم كل 2-4 أسابيع، وتُحسب كمية الدم بناءً على الوزن (عادةً 10-15 مل من كريات الدم الحمراء المركّزة لكل كيلوغرام من وزن الجسم في كل جلسة). قبل كل عملية نقل تُجرى اختبارات التوافق (Cross-matching) للتأكد من عدم وجود أجسام مضادة قد تسبّب تفاعلات نقل الدم.
التحديات تشمل: تطوّر أجسام مضادة ضد كريات الدم المنقولة (Alloimmunization) مما يُصعّب إيجاد دم متوافق مستقبلاً؛ وخطر انتقال العدوى (رغم أن أنظمة فحص الدم الحديثة في السعودية قلّلت هذا الخطر بشدة)؛ وبالطبع تراكم الحديد.
العلاج بالاستخلاب: طرد الحديد الزائد من الجسم
⚠️ تحذير: أدوية الاستخلاب تتطلب جرعات دقيقة ومتابعة مخبرية مستمرة. الجرعات المذكورة هنا إرشادية ويجب تعديلها من قبل طبيب أمراض الدم المعالج.
أدوية الاستخلاب (Iron Chelation Therapy) هي “المكنسة” التي تجمع الحديد الزائد من أنسجة الجسم وتطرحه عبر البول أو البراز. وهي ضرورة حياتية لكل مريض يخضع لنقل دم منتظم. هناك ثلاثة أدوية رئيسة:
ديفيروكسامين (Deferoxamine — Desferal®): أقدم أدوية الاستخلاب وأكثرها خبرة سريرية. يُعطى عادةً عبر التسريب تحت الجلد (Subcutaneous Infusion) بجرعة 20-40 ملغ/كغ/يوم لمدة 8-12 ساعة يومياً (5-7 أيام أسبوعياً). الأطفال: يبدأ عادةً بعد 10-20 عملية نقل دم أو عندما يتجاوز الفيريتين 1000 نانوغرام/مل. تبدأ الجرعة عادةً بـ 20-30 ملغ/كغ ثم تُعدَّل. البالغون: 30-50 ملغ/كغ/يوم. الحوامل: يُعَدّ الديفيروكسامين غير آمن في الثلث الأول من الحمل (فئة C). يُوقف عادةً قبل الحمل المخطط ولا يُستأنف إلا إذا كانت الحاجة ملحّة جداً وبموافقة فريق التوليد وأمراض الدم معاً. المرضعات: لا توجد بيانات كافية عن إفرازه في حليب الأم؛ يُوصى بالحذر واستشارة الطبيب. كبار السن: تُعدَّل الجرعة بحسب وظائف الكلى والسمع والنظر.
الآثار الجانبية: تفاعلات موضعية في مكان الحقن (تورّم، احمرار)؛ سُمّية سمعية (فقدان سمع حسّي عصبي) خاصة عند الأطفال بجرعات مرتفعة؛ سُمّية بصرية (اضطرابات في شبكية العين)؛ تأخر نمو العظام عند الأطفال إذا كانت الجرعة أعلى من 40 ملغ/كغ. فرط الجرعة: قد يسبب هبوطاً حاداً في ضغط الدم وتسرّع القلب وفشلاً كلوياً حاداً. في حالة الاشتباه بالجرعة المفرطة، يجب التوجه للطوارئ فوراً.
ديفيريبرون (Deferiprone — Ferriprox®): يؤخذ عن طريق الفم. الجرعة المعتادة: 75-100 ملغ/كغ/يوم مقسّمة على 3 جرعات يومية. للأطفال فوق 6 سنوات والبالغين. يتميّز بقدرته العالية على إزالة الحديد من عضلة القلب تحديداً، ولذلك يُفضّل في حالات تراكم الحديد القلبي. الأثر الجانبي الأخطر: ندرة المحبّبات (Agranulocytosis) — أي انخفاض حاد في كريات الدم البيضاء قد يُعرّض المريض لعدوى خطيرة. لذلك يجب إجراء تعداد كريات بيضاء أسبوعياً. أعراض أخرى تشمل: آلام المفاصل، غثيان، ارتفاع إنزيمات الكبد. الحوامل والمرضعات: ممنوع استخدامه (فئة X — ثبتت سُمّيته الجنينية في الدراسات الحيوانية).
ديفيراسيروكس (Deferasirox — Exjade® / Jadenu®): الأحدث والأكثر ملاءمة لنمط الحياة اليومي لأنه يؤخذ عن طريق الفم مرة واحدة يومياً. الجرعة: أقراص Jadenu®: 14-28 ملغ/كغ/يوم (تؤخذ مع الطعام أو بدونه). أقراص Exjade® القابلة للتشتيت في الماء: 20-40 ملغ/كغ/يوم على معدة فارغة قبل الطعام بـ 30 دقيقة. للأطفال من عمر سنتين فما فوق والبالغين. الأعراض الجانبية: اضطرابات هضمية (غثيان، إسهال، ألم بطن)؛ ارتفاع الكرياتينين (مؤشر على تأثّر الكلى)؛ ارتفاع إنزيمات الكبد. يتطلّب فحص وظائف الكلى والكبد شهرياً. فرط الجرعة: قد يؤدي لفشل كلوي حاد وفشل كبدي.
كبار السن وأصحاب الأمراض المزمنة: تُعدَّل جرعات جميع أدوية الاستخلاب بحسب وظائف الكلى والكبد. مرضى السكري يحتاجون مراقبة أدق لوظائف الكلى. مرضى القلب يحتاجون تنسيقاً وثيقاً بين طبيب الدم وطبيب القلب.
| المعيار | ديفيروكسامين (Desferal®) | ديفيريبرون (Ferriprox®) | ديفيراسيروكس (Jadenu®) |
|---|---|---|---|
| طريقة الإعطاء | تسريب تحت الجلد (8-12 ساعة) | فم — 3 جرعات يومياً | فم — جرعة واحدة يومياً |
| الجرعة المعتادة للبالغين | 30-50 ملغ/كغ/يوم | 75-100 ملغ/كغ/يوم | 14-28 ملغ/كغ/يوم (Jadenu®) |
| ميزة رئيسية | أكثر خبرة سريرية وأقدم | فعّال جداً لاستخلاب حديد القلب | الأسهل استخداماً يومياً |
| الأثر الجانبي الأخطر | سُمّية سمعية وبصرية | ندرة المحبّبات (Agranulocytosis) | تأثر الكلى والكبد |
| المتابعة الضرورية | سمع، نظر، نمو العظام | تعداد كريات بيضاء أسبوعياً | وظائف كلى وكبد شهرياً |
| الأمان في الحمل | يُوقف في الثلث الأول (فئة C) | ممنوع تماماً (فئة X) | يُوقف — بيانات غير كافية |
| الحد الأدنى للعمر | بعد 10-20 نقل دم | 6 سنوات فأكثر | سنتان فأكثر |
يوصي المستشار الدوائي جاسم محمد مراد — خبير الصحة والإمداد الطبي في موقع وصفة طبية — بضرورة “عدم التبديل بين أدوية الاستخلاب أو تعديل جرعاتها ذاتياً أبداً. كل دواء له ملف أمان مختلف وآثار جانبية نوعية يجب مراقبتها بفحوصات محددة. المريض الذي يشعر بأعراض جانبية لا يوقف الدواء — بل يتصل بطبيبه ليقرر البديل.”
المكملات الغذائية الضرورية
⚠️ تحذير: المكملات التالية لا تغني عن العلاج الطبي الأساسي، ويجب تناولها بإشراف طبي.
حمض الفوليك (Folic Acid): ضروري لمرضى الثلاسيميا لأن نخاع العظم المُجهَد يستهلك كميات كبيرة منه في محاولته المتكررة لتصنيع كريات دم جديدة. الجرعات المعتمدة:
- الأطفال (1-12 سنة): 1 ملغ يومياً.
- البالغون (أكبر من 12 سنة): 1-5 ملغ يومياً.
- الحوامل المصابات: 5 ملغ يومياً (الجرعة الأعلى ضرورية لتقليل خطر عيوب الأنبوب العصبي في الجنين).
- المرضعات: 1 ملغ يومياً.
الآثار الجانبية نادرة وتشمل: غثيان خفيف، انتفاخ، وفي حالات نادرة جداً حساسية. حمض الفوليك آمن عموماً حتى بجرعات عالية نسبياً.
فيتامين D والكالسيوم: بسبب ارتفاع خطر هشاشة العظام. الجرعة تُحدَّد بناءً على مستوى فيتامين D في الدم:
- الأطفال: 600-1000 وحدة دولية يومياً من فيتامين D3، مع 500-1000 ملغ كالسيوم حسب العمر.
- البالغون: 1000-2000 وحدة دولية يومياً (أو أكثر في حالات النقص الشديد بإشراف طبي)، مع 1000-1200 ملغ كالسيوم.
- كبار السن: نفس الجرعة مع مراقبة مستوى الكالسيوم في الدم لتجنّب فرط الكالسيوم.
- الحوامل: 600-1000 وحدة دولية يومياً.
الزنك (Zinc): ينخفض مستواه عند كثير من مرضى الثلاسيميا بسبب أدوية الاستخلاب (خاصة الديفيروكسامين). الجرعة: 15-25 ملغ يومياً للبالغين، 5-10 ملغ للأطفال. يؤخذ بعيداً عن مكملات الحديد والكالسيوم بساعتين على الأقل.
تحذير مهم بخصوص الحديد: يُمنع تماماً تناول مكملات الحديد لمرضى الثلاسيميا الكبرى والوسطى إلا في حالات استثنائية نادرة يحددها الطبيب بناءً على فحص مخزون الحديد. حتى حاملو السمة يجب أن يتأكدوا من وجود نقص حديد حقيقي (عبر فحص الفيريتين والحديد المصلي) قبل تناول أي مكمل حديد.
بخصوص المكملات العشبية: يسأل كثير من المرضى عن الكركم والشاي الأخضر والحلبة. دعونا نكون صريحين:
- الكركم (الكركمين): يملك خصائص مضادة للأكسدة نظرياً، لكن المكملات المركّزة قد تتداخل مع أدوية الاستخلاب (خاصة ديفيراسيروكس) وقد تزيد من خطر النزيف عند المرضى الذين يتناولون مضادات التخثر. استخدامه كتوابل طبخ عادية آمن. أما المكملات المركّزة فلا تبدأها دون استشارة طبيبك.
- الشاي الأخضر: يحتوي على مواد (الكاتيكينات) قد تُقلّل امتصاص الحديد من الطعام — وهذا قد يكون مفيداً نظرياً لمرضى الثلاسيميا. لكن لا توجد دراسات سريرية كافية لتوصيته كعلاج مساعد. شربه باعتدال (كوب إلى كوبين يومياً) آمن عموماً ولا يتعارض مع أدوية الاستخلاب بجرعات عادية.
- الحلبة (Trigonella foenum-graecum): لا يوجد دليل علمي قوي على فائدتها لمرضى الثلاسيميا. قد تخفّض سكر الدم لدى مرضى السكري المصاحب، مما يتطلب تعديل جرعة أدوية السكر. آمنة بالجرعات الغذائية.
يؤكّد المستشار الدوائي جاسم محمد مراد أن “القاعدة الذهبية لأي مريض ثلاسيميا: لا تُضف أي مكمل غذائي أو عشبي إلى قائمة أدويتك قبل مراجعة طبيبك أو الصيدلي السريري. بعض المكملات التي تبدو بريئة قد تتداخل مع أدوية الاستخلاب بطرق تُقلّل فعاليتها أو تزيد سُمّيتها.”
زراعة نخاع العظم أو الخلايا الجذعية: العلاج الجذري
زراعة الخلايا الجذعية المكوّنة للدم (Hematopoietic Stem Cell Transplantation — HSCT) هي العلاج الوحيد الشافي حالياً لمرض الثلاسيميا الكبرى. الفكرة ببساطة: نستبدل نخاع العظم المعيب بنخاع سليم من متبرع متوافق.
أفضل النتائج تتحقق عندما يكون المتبرع أخاً أو أختاً شقيقاً متطابقاً في نظام HLA (Human Leukocyte Antigen)، وعندما يكون عمر المريض صغيراً (أقل من 14 عاماً) ولم يتطور لديه تليّف كبدي أو تضخم طحال شديد. نسبة الشفاء من مرض الثلاسيميا بالزراعة في هذه الظروف المثالية تتجاوز 85-90%.
التحديات: ليس كل مريض يجد متبرعاً متوافقاً (الاحتمال 25% فقط لكل شقيق). مخاطر الزراعة تشمل رفض الطعم (Graft Rejection)، ومرض الطعم ضد المضيف (Graft-versus-Host Disease — GVHD)، ومضاعفات العلاج الكيميائي التحضيري. في السعودية، تتوفر خدمات زراعة النخاع في عدة مراكز متقدمة مثل مستشفى الملك فيصل التخصصي ومركز الأبحاث بالرياض وجدة.
تشير الدكتورة سوزان عبد الحميد السعدي — استشارية أمراض الدم وزرع الخلايا الجذعية في موقع وصفة طبية — إلى أن “التوقيت المثالي للزراعة هو قبل حدوث مضاعفات تراكم الحديد الخطيرة. كلما كان المريض أصغر سناً وأقل تحمّلاً بالحديد، كانت نتائج الزراعة أفضل بمراحل. ولذلك ننصح أسر الأطفال المصابين بمناقشة خيار الزراعة مبكراً مع فريقهم الطبي.”
العلاجات الجينية الحديثة: نافذة أمل جديدة
هذا هو الجزء الذي يُضيء المستقبل. في عام 2023 وافقت إدارة الغذاء والدواء الأميركية (FDA) على أول علاج جيني للثلاسيميا المعتمدة على نقل الدم: عقار Casgevy® (Exagamglogene Autotemcel) الذي يستخدم تقنية كريسبر-كاس 9 (CRISPR-Cas9) لتعديل الخلايا الجذعية الخاصة بالمريض نفسه، بحيث تُعيد تفعيل إنتاج الهيموغلوبين الجنيني (HbF) الذي يعوّض النقص في الهيموغلوبين البالغ.
كذلك وافقت FDA على عقار Zynteglo® (Betibeglogene Autotemcel) الذي يستخدم تقنية العلاج الجيني بالنواقل الفيروسية (Lentiviral Vector Gene Therapy) لإدخال نسخة وظيفية من جين بيتا غلوبين إلى الخلايا الجذعية للمريض. النتائج الأولية أظهرت أن نسبة كبيرة من المرضى المعالجين استغنوا تماماً عن نقل الدم لسنوات بعد العلاج.
هل مرض الثلاسيميا خطير في ضوء هذه التطورات؟ بالتأكيد لا يزال مرضاً جدّياً يحتاج متابعة دقيقة. لكنّ المشهد العلاجي تغيّر جذرياً. لم يعد السؤال “هل يوجد شفاء؟” بل أصبح “متى يصل هذا العلاج إلى كل المرضى؟”
من المثير أن تعرف: تكلفة عقار Zynteglo® في الولايات المتحدة تبلغ نحو 2.8 مليون دولار أميركي لجرعة واحدة، مما يجعله من أغلى الأدوية في تاريخ الطب. لكن حين تُحسب تكلفة نقل الدم والاستخلاب مدى الحياة (تتجاوز 6 ملايين دولار في بعض التقديرات)، يبدو العلاج الجيني مجدياً اقتصادياً على المدى الطويل.
كيف يبدو روتين الحياة والتعايش السليم مع الثلاسيميا؟
النظام الغذائي المثالي: ماذا نأكل وماذا نتجنّب؟

⚠️ تحذير: النصائح الغذائية التالية لا تُغني عن المتابعة مع اختصاصي تغذية مطّلع على حالة المريض.
الأكل الممنوع لمرضى الثلاسيميا — أو الأدقّ: الأطعمة التي يجب تقليلها — هي تلك الغنية بالحديد سهل الامتصاص (حديد الهيم — Heme Iron) الموجود في اللحوم الحمراء والكبد والطحال. هذا لا يعني الامتناع التام عن اللحوم؛ بل يعني التقليل الذكي وعدم الإفراط.
على النقيض من ذلك، هناك أطعمة تُقلّل امتصاص الحديد من الأمعاء ويمكن استغلالها بذكاء:
- الشاي والقهوة: شربها مع الوجبات (وليس بعيداً عنها) يُقلّل امتصاص الحديد النباتي.
- الأطعمة الغنية بالكالسيوم: الحليب والجبن واللبن — تتنافس مع الحديد على الامتصاص في الأمعاء.
- الحبوب الكاملة والبقوليات: تحتوي على حمض الفيتيك (Phytic Acid) الذي يُعيق امتصاص الحديد.
الأطعمة المطلوبة: الخضراوات الورقية (لحمض الفوليك)، الفواكه الغنية بمضادات الأكسدة (التوت، الرمان)، الأسماك (لأحماض أوميغا-3 المضادة للالتهاب وفيتامين D).
تنصح الدكتورة علا الأحمد — اختصاصية التغذية العلاجية في موقع وصفة طبية — بأن “الهدف ليس حرمان المريض من الطعام، بل بناء نظام غذائي ذكي يُقلّل الحمل الحديدي من المصادر الغذائية دون التسبب في نقص عناصر أخرى. أنصح بالتركيز على وجبات متوازنة تحتوي على بروتينات نباتية وخضراوات ملوّنة، مع شرب كوب شاي أو قهوة مع الوجبة الرئيسة لتقليل امتصاص الحديد.”
اقرأ أيضاً:
ممارسة الرياضة بأمان
مريض الثلاسيميا يمكنه ممارسة الرياضة — بل يُشجَّع على ذلك — لكن بشروط. الرياضات المعتدلة كالمشي والسباحة وركوب الدراجة مناسبة جداً. الرياضات العنيفة التي تُعرّض البطن للإصابة (ككرة القدم والفنون القتالية) يجب تجنّبها إذا كان الطحال متضخّماً، خوفاً من التمزّق.
المهم هو أن يستمع المريض لجسمه: إذا شعر بضيق تنفّس أو إرهاق شديد في أثناء التمرين، فعليه التوقف والراحة. ويُفضَّل ممارسة الرياضة بعد عملية نقل الدم بيوم أو يومين حين يكون الهيموغلوبين في أعلى مستوياته.
الحماية من العدوى: أهمية التطعيمات
المرضى الذين خضعوا لاستئصال الطحال معرّضون بشدة للعدوى البكتيرية الخطيرة. التطعيمات التالية ضرورية:
- لقاح المكوّرات الرئوية (Pneumococcal Vaccine — PCV13 + PPSV23).
- لقاح المستدمية النزلية نوع ب (Haemophilus influenzae type b — Hib).
- لقاح السحائيات (Meningococcal Vaccine).
- لقاح الإنفلونزا السنوي.
بالإضافة إلى ذلك، يُوصف للمرضى بعد استئصال الطحال مضاد حيوي وقائي (بنسلين V أو أموكسيسيلين) يومياً — في كثير من الحالات مدى الحياة — وهذا ليس ترفاً بل ضرورة.
الثلاسيميا في المدرسة: كيف تدعم طفلك أكاديمياً؟
الغياب المتكرر بسبب مواعيد نقل الدم أو الإرهاق قد يؤثر على التحصيل الدراسي للطفل. الحل يكمن في التواصل الاستباقي الشفاف مع إدارة المدرسة:
- احرص على تقديم تقرير طبي مفصل في بداية العام الدراسي لإدارة المدرسة والمرشد الطلابي، يشرح حالة الطفل بوضوح.
- اطلب استثناءات بسيطة ولكنها جوهرية: كالسماح للطفل بشرب الماء بكثرة أثناء الحصص، وأخذ فترات راحة قصيرة إذا شعر بالإرهاق، وإعفائه من التمارين الرياضية القاسية في حصص التربية البدنية.
- تعاون مع المعلمين لجدولة الواجبات أو الامتحانات التي قد تتزامن مع أيام نقل الدم التي يكون فيها الطفل في أدنى مستويات طاقته.
مرضى الثلاسيميا في بيئة العمل: حقوق وإنتاجية
بالنسبة للبالغين، إخفاء المرض في بيئة العمل قد يزيد من الضغط النفسي والجسدي. الموظف المصاب بالثلاسيميا هو شخص منتج وطبيعي، لكنه يحتاج إلى بيئة عمل مرنة (Flexible Workplace). يُنصح بمصارحة قسم الموارد البشرية (HR) أو المدير المباشر بالحالة لضمان تفهمهم عند الحاجة لأخذ إجازة مرضية دورية لنقل الدم، أو لجدولة مواعيد العمل بما يتناسب مع المراجعات الطبية الدورية، مما يضمن استمرارية الإبداع والإنتاجية دون مساومة على الصحة.
هل تعلم؟ في دراسة منشورة في The Lancet عام 2022 تبيّن أن مرضى الثلاسيميا بعد استئصال الطحال الذين لم يتلقوا مضاداً حيوياً وقائياً كانوا أكثر عرضة بـ 50 ضعفاً للإصابة بتسمّم دم مميت مقارنة بمن التزموا بالعلاج الوقائي.
اقرأ أيضاً:
هل يُشير فقر الدم المزمن إلى أمراض أخرى في الجسم؟
فقر الدم الذي يرافق الثلاسيميا ليس بالضرورة الصورة الوحيدة لنقص الأكسجة النسيجية المزمنة في هذا المرض. كما أن بعض مضاعفات الثلاسيميا تتشابه مع أمراض جهازية أخرى يجب استبعادها أو مراقبتها:
السكري (Diabetes Mellitus): تراكم الحديد في خلايا بيتا البنكرياسية (Pancreatic β-cells) يُعطّل إفراز الأنسولين، مما يسبب ما يُعرف بالسكري الثانوي لتراكم الحديد (Iron-Overload Diabetes). يُوصى بفحص الغلوكوز التراكمي (HbA1c) سنوياً لمرضى الثلاسيميا — مع الأخذ بعين الاعتبار أن قراءة HbA1c قد تكون غير دقيقة عندهم بسبب نقل الدم المتكرر، ولذلك يُفضَّل اختبار الغلوكوز الصيامي أو اختبار تحمّل الغلوكوز الفموي (OGTT).
قصور الغدة الدرقية (Hypothyroidism): ترسّب الحديد في الغدة الدرقية يؤدي تدريجياً إلى قصورها. الأعراض (خمول، زيادة وزن، إمساك) قد تتداخل مع أعراض فقر الدم نفسه، لذلك يجب فحص TSH وT4 دورياً.
قصور الغدد جارات الدرقية (Hypoparathyroidism): يسبب انخفاض الكالسيوم في الدم، مما يؤدي لتشنّجات عضلية وتنميل حول الفم وأحياناً نوبات صرعية.
هشاشة العظام (Osteoporosis): كما ذكرنا، ناتجة عن عدة آليات متداخلة (فرط نشاط النخاع، نقص هرمونات، نقص فيتامين D والكالسيوم). قد تكون هشاشة العظام عند شاب في العشرين مؤشراً على خلل هرموني ثانوي لتراكم الحديد يجب البحث عنه.
اقرأ أيضاً:
- مقاومة الإنسولين: القاتل الصامت وكيفية عكسه لاستعادة صحتك الجسدية
- السكري من النوع الثاني: الدليل الشامل لفهم المرض، السيطرة عليه، وتجنب مضاعفاته
الثلاسيميا والحمل والإرضاع: المسموح والممنوع
⚠️ تحذير أساسي: يجب على كل امرأة مصابة بالثلاسيميا (سواء الوسطى أو الكبرى) أن تُخطّط لحملها مسبقاً مع فريق طبي يضمّ اختصاصي أمراض دم وطبيب توليد عالي الخطورة، ولا تحمل قبل ضبط مخزون الحديد وتقييم وظائف القلب.
المسموح والآمن نسبياً:
- حمض الفوليك (5 ملغ يومياً) ضروري قبل الحمل وخلاله.
- نقل الدم للحفاظ على هيموغلوبين أعلى من 10 غ/دل خلال الحمل.
- فيتامين D والكالسيوم بالجرعات الموصى بها.
- المراقبة القلبية الدورية بالإيكو (Echocardiography) كل ثلاثة أشهر.
الممنوع:
- ديفيريبرون (Deferiprone): ممنوع تماماً — ثبتت سُمّيته الجنينية. يجب إيقافه قبل الحمل بثلاثة أشهر على الأقل.
- ديفيراسيروكس (Deferasirox): لا توجد بيانات أمان كافية في البشر. يُوقف قبل الحمل.
- ديفيروكسامين (Deferoxamine): الأقل خطورة نسبياً، لكنه يُوقف في الثلث الأول من الحمل. قد يُستأنف في الثلثين الثاني والثالث إذا كان تراكم الحديد يهدد حياة الأم (خاصة الحديد القلبي) — والقرار يُتّخذ بمشاركة فريق طبي متعدد التخصصات.
الآلية الفسيولوجية للضرر: أدوية الاستخلاب ترتبط بالحديد وبالمعادن الأخرى الضرورية لنمو الجنين (الزنك والنحاس)، كما قد تعبر المشيمة وتؤثر على تكوّن أعضاء الجنين في المراحل المبكرة.
في أثناء الإرضاع: لا توجد بيانات كافية عن إفراز أدوية الاستخلاب في حليب الأم. يُفضّل عدم استخدامها في أثناء الرضاعة إلا عند الضرورة القصوى، والقرار يعود للفريق الطبي.
اقرأ أيضاً:
الثلاسيميا عند الأطفال: ملاحظات خاصة
الأطفال هم الفئة الأكثر تأثراً بالثلاسيميا الكبرى، وتبدأ رحلتهم العلاجية من السنة الأولى. بعض الملاحظات الخاصة:
النمو والتطور: يجب مراقبة الوزن والطول ومعالم التطور الحركي والذهني في كل زيارة. التأخر يستدعي تقييماً هرمونياً شاملاً.
البلوغ: تأخر البلوغ شائع بسبب ترسّب الحديد في الغدة النخامية والغدد التناسلية. قد يحتاج الطفل لعلاج هرموني تعويضي (هرمونات البلوغ) بإشراف اختصاصي غدد صمّاء أطفال.
التطعيمات: نفس جدول التطعيمات الوطني، مع إضافة اللقاحات الخاصة بمنزوعي الطحال إذا استُؤصل.
الصحة النفسية: الطفل الذي يتردد على المستشفى كل أسبوعين أو ثلاثة أسابيع لنقل الدم يتعرّض لضغط نفسي كبير. الدعم النفسي والمشاركة في أنشطة اجتماعية وأقران يعانون من نفس الحالة يُحسّن جودة حياته كثيراً. لا تتجاهل الشكوى النفسية لطفلك؛ اطلب تقييماً نفسياً إذا لاحظت انسحاباً أو حزناً مستمراً.
حقيقة طبية: أثبتت دراسة منشورة في Journal of Pediatric Hematology/Oncology عام 2021 أن الأطفال المصابين بالثلاسيميا الكبرى الذين يتلقون دعماً نفسياً منتظماً بالتوازي مع العلاج الطبي يُظهرون التزاماً أفضل بأدوية الاستخلاب ونتائج صحية أفضل على المدى الطويل.
رعاية مقدم الرعاية: الأكسجين لك أولاً أيها الأب / أيتها الأم
غالباً ما ينسى الآباء والأمهات أنفسهم في زحمة المواعيد الطبية، وجلسات نقل الدم، ومراقبة مضاعفات الأدوية لأطفالهم. إذا كنت الوالد أو مقدم الرعاية الأساسي لطفل مصاب بالثلاسيميا، فاعلم أن “احتراق مقدم الرعاية” (Caregiver Burnout) ليس ضعفاً منك، بل هو نتيجة طبيعية للضغط المستمر.
القاعدة الذهبية في الطوارئ الجوية تنطبق هنا: “ضع قناع الأكسجين لنفسك أولاً قبل مساعدة طفلك”. لا يمكنك تقديم رعاية ممتازة لطفلك إذا كنت مستنزفاً جسدياً ونفسياً.
- اطلب المساعدة: تبادل الأدوار مع شريك حياتك أو أفراد العائلة الموثوقين في مرافقة الطفل للمستشفى.
- احمِ وقتك الخاص: خصص ولو 30 دقيقة يومياً للابتعاد عن التفكير في المرض؛ سواء للمشي، أو القراءة، أو مجرد الاسترخاء.
- تحدث عن مخاوفك: الانضمام إلى مجموعات دعم أهالي مرضى الثلاسيميا يمنحك مساحة للتنفيس مع أشخاص يعيشون نفس تجربتك ويفهمون تماماً معنى أن تسهر ليلاً تراقب مضخة الاستخلاب.
اقرأ أيضاً:
- رفض الرضيع للثدي: الأسباب الخفية والحلول العملية لإنقاذ رحلة الرضاعة
- تأخر الكلام واللغة عند الأطفال: الأسباب الطبية والعلامات التحذيرية وأحدث طرق العلاج
كبار السن ومرضى الثلاسيميا: تحديات مضاعفة
مع تحسّن الرعاية الطبية، أصبح عدد متزايد من مرضى الثلاسيميا يصلون إلى مرحلة الشيخوخة — وهذا إنجاز طبي حقيقي، لكنه يطرح تحديات جديدة. كبار السن المصابون بالثلاسيميا يعانون من تراكم المضاعفات المزمنة: فشل قلبي، سكري، قصور غدد صمّاء متعدد، هشاشة عظام شديدة. تعديل جرعات أدوية الاستخلاب يصبح أكثر حساسية بسبب تراجع وظائف الكلى والكبد مع التقدم في العمر. المتابعة الدورية الشاملة (قلب، غدد، كبد، كلى، عظام) كل 3-6 أشهر ضرورية. كما أن التداخلات الدوائية تصبح أكثر تعقيداً حين يتناول المريض أدوية لأمراض مزمنة أخرى (ضغط، سكري، كوليسترول) إلى جانب أدوية الاستخلاب.
فحص ما قبل الزواج: الدرع الواقي للأجيال القادمة
في السعودية، يُعَدّ فحص الثلاسيميا قبل الزواج إلزامياً منذ عام 2004 ضمن برنامج الفحص الصحي لما قبل الزواج (Premarital Screening Program) الذي يشمل أيضاً فقر الدم المنجلي. البرنامج لا يمنع الزواج قانونياً حتى لو كان الطرفان حاملين، لكنه يُلزم بإبلاغهما بالمخاطر ليتّخذا قرارهما بوعي.
فما فائدة هذا الفحص عملياً؟ منذ تطبيق البرنامج، انخفضت نسبة الزيجات عالية الخطورة (بين حاملين اثنين) من نحو 10% إلى أقل من 5% في بعض المناطق، وانخفض عدد المواليد المصابين بأشكال شديدة من الثلاسيميا. لكنّ التحدي يبقى في أن بعض الأزواج يختارون المضي رغم النتيجة، إما بسبب ضغوط عائلية أو عدم فهم كافٍ للمخاطر.
النصيحة المباشرة: إذا كنت مقبلاً على الزواج — أجرِ فحص الثلاسيميا قبل الزواج حتى لو لم تكن في منطقة ينتشر فيها المرض. الفحص بسيط (سحب دم)، ونتيجته قد تحمي أطفالك من معاناة يمكن تجنّبها.
كم تكلّف علاجات الثلاسيميا تقريباً؟
الشفافية المالية جزء من الوعي الطبي. إليك تقديرات تقريبية:
نقل الدم الدوري: في المستشفيات الحكومية السعودية يُقدَّم مجاناً ضمن التأمين الصحي الحكومي. في المستشفيات الخاصة، تتراوح تكلفة الجلسة الواحدة (شاملة الدم والتوافق والتمريض) بين 1,500 و4,000 ريال سعودي (400-1,000 دولار أميركي).
أدوية الاستخلاب: ديفيراسيروكس (Jadenu®) قد يكلّف 3,000-8,000 ريال شهرياً (800-2,100 دولار) حسب الجرعة ومصدر الدواء. في السعودية، تُغطّي وزارة الصحة هذه الأدوية للمرضى المسجّلين في مراكز الثلاسيميا الحكومية.
زراعة نخاع العظم: تتراوح التكلفة بين 150,000 و500,000 ريال (40,000 – 130,000 دولار) في السعودية حسب المركز ونوع المتبرع ومضاعفات ما بعد الزراعة. عالمياً قد تصل إلى 300,000 دولار.
العلاج الجيني (Casgevy® أو Zynteglo®): غير متوفر حالياً في السعودية، وتكلفته في أميركا تتراوح بين 2.2 و2.8 مليون دولار للجرعة الواحدة.
العوامل التي تتحكّم في التكلفة: نوع المركز الطبي (حكومي أو خاص)، مدى توفّر التأمين الصحي، شدة المرض وعدد مرات نقل الدم، وجود مضاعفات تتطلّب تدخلات إضافية (قلب، غدد، عظام).
الخطة العملية للتعامل مع مرض الثلاسيميا يومياً
- التزم بمواعيد نقل الدم بدقة — لا تؤجّل الموعد أبداً. ضع تذكيراً في هاتفك قبل الموعد بيومين لتحضير نفسك والملف الطبي.
- تناول أدوية الاستخلاب في نفس الوقت يومياً — الثبات في التوقيت يُحسّن الالتزام ويُقلّل نسيان الجرعات. إذا نسيت جرعة، تناولها فور تذكّرك ما لم يكن قريباً من موعد الجرعة التالية.
- احتفظ بسجل مخبري شخصي — سجّل فيه مستوى الهيموغلوبين قبل وبعد كل عملية نقل، والفيريتين كل 3 أشهر، ونتائج الرنين المغناطيسي السنوية. هذا السجل يُسهّل على أي طبيب جديد فهم حالتك بسرعة.
- أجرِ تقييماً قلبياً شاملاً مرة سنوياً على الأقل — إيكو القلب + رنين مغناطيسي T2* للقلب. لا تنتظر ظهور أعراض.
- اخضع لفحوصات الغدد الصمّاء سنوياً — تشمل: وظائف الدرقية، غلوكوز الدم الصيامي، هرمونات البلوغ (عند المراهقين)، كالسيوم الدم وفيتامين D.
- لا تتناول أي مكمل حديد أبداً دون فحص مخبري يُثبت نقصاً حقيقياً (وهذا نادر جداً في مرضى الثلاسيميا المعتمدين على نقل الدم).
- تواصل مع مجموعات دعم المرضى — في السعودية توجد جمعيات خيرية لدعم مرضى الثلاسيميا تُقدّم مساعدات مادية وتوعوية ونفسية.
- احذر فخ “تمرد المراهقة” عند انتقال الرعاية: تُعد مرحلة المراهقة والانتقال من عيادات طب الأطفال إلى عيادات البالغين (Transition of Care) من أخطر المراحل. في هذا العمر، قد يميل المراهق إلى إنكار المرض، أو التمرد على أدوية الاستخلاب، أو الشعور بالخجل من مضاعفات المرض أمام أقرانه. إذا كنت والداً لمراهق، ابدأ بنقل مسؤولية إدارة المرض إليه تدريجياً (مثل متابعة مواعيد الأدوية بنفسه) مع بقائك مراقباً وداعماً. وإذا كنت أنت المريض الشاب؛ فتذكر أن التزامك الآن هو ما سيرسم جودة حياتك في العشرينيات والثلاثينيات.
- احمل بطاقة طبية دائماً — مكتوب فيها تشخيصك، أدويتك، فصيلة دمك، وحالة الطحال (موجود أم مستأصل). في حالة الطوارئ، هذه البطاقة قد تُنقذ حياتك.
الوصفة الطبية من موقعنا
- ركّز على الأطعمة الغنية بمضادات الأكسدة الفينولية: التوت البري والرمان والزيتون تحتوي على مركبات بوليفينولية (Polyphenols) تعمل على معادلة الشوارد الحرة (Free Radicals) التي يُولّدها الحديد الزائد عبر تفاعل فنتون (Fenton Reaction) داخل الخلايا. هذا لا يحلّ محل الاستخلاب، لكنه يُقلّل الضرر التأكسدي اليومي.
- اشرب الشاي الأسود أو الأخضر مع وجباتك الرئيسة: التانينات (Tannins) والكاتيكينات (Catechins) في الشاي ترتبط بالحديد النباتي في الأمعاء وتُقلّل امتصاصه بنسبة تصل إلى 60% — وفق دراسة منشورة في American Journal of Clinical Nutrition. هذه خطوة بسيطة لكنها فعّالة.
- حافظ على نوم 7-8 ساعات في ظلام تام: الميلاتونين (Melatonin) — الذي يُفرز أثناء النوم في الظلام — يملك خصائص مضادة للأكسدة ومحمية للكبد. أي اضطراب في الإيقاع اليوماوي (Circadian Rhythm) يُضعف قدرة الكبد على التعامل مع الحمل الحديدي.
- مارس المشي المعتدل 20-30 دقيقة يومياً بعد نقل الدم بيوم: النشاط البدني الخفيف يُحسّن تدفّق الدم ويرفع كفاءة إيصال الأكسجين للأنسجة. كما أنه يُنشّط إفراز عامل النمو الشبيه بالأنسولين (IGF-1) الذي يدعم صحة العظام — وهي نقطة حرجة لمرضى الثلاسيميا.
- قلّل من استهلاك فيتامين C كمكمّل مُركّز: فيتامين C يزيد امتصاص الحديد من الأمعاء بقوة. تناوله بكميات الطعام الطبيعية آمن، لكن تجنّب أقراص الفوّار عالية الجرعة (1000 ملغ) إلا إذا أخذتها مع جرعة الاستخلاب تحديداً بإشراف طبي (بعض البروتوكولات تستخدم جرعة صغيرة من فيتامين C مع ديفيروكسامين لتعزيز طرح الحديد — لكن فقط بإشراف طبي صارم).
- ضع خطة لإدارة التوتر: الإجهاد النفسي المزمن يرفع مستويات الكورتيزول، الذي يُضعف المناعة ويُسرّع ارتشاف العظام (Bone Resorption). تقنيات بسيطة كالتنفس العميق والتأمل لمدة 10 دقائق يومياً أظهرت أثراً قابلاً للقياس في تحسين المؤشرات الالتهابية لدى مرضى الأمراض المزمنة.
اقرأ أيضاً:
خاتمة ورسالة دعم
مرض الثلاسيميا ليس نهاية الطريق. نعم، هو مرض مزمن يتطلب صبراً والتزاماً ومتابعة طبية دقيقة. لكنّ العلم تقدّم بشكل مذهل خلال العقود الأخيرة: من أدوية استخلاب أكثر فعالية، إلى زراعة نخاع عظم بنسب نجاح مرتفعة، إلى علاجات جينية تُبشّر بشفاء حقيقي. المريض الذي يلتزم بخطته العلاجية، ويُجري فحوصاته الدورية، ويتغذى بذكاء، ويحافظ على معنوياته — يمكنه أن يعيش حياة طويلة ومنتجة وسعيدة.
لكل أب وأم لطفل مصاب: أنتم لستم وحدكم. هناك فرق طبية متخصصة في السعودية والخليج والعالم العربي تعمل يومياً لتحسين حياة أطفالكم. ولكل مراهق أو شاب يحمل هذا التشخيص: مرضك لا يُحدّد هويتك. أنت أكبر من تشخيص طبي.
هل أجريت فحص الثلاسيميا قبل الزواج لك ولشريكك؟ إن لم تكن قد فعلت — فهذا هو أول شيء يجب أن تفعله اليوم.
اقرأ أيضاً:
- الطب الشعوري التصنيفي: خريطة الشفاء عبر فك شيفرة المشاعر والأمراض المكبوتة
- صندوق الإسعافات الأولية: كيف تنقذ حياة من تحب في اللحظات الحرجة؟
أسئلة شائعة عن مرض الثلاسيميا
✅ بيان المصداقية — موقع وصفة طبية
يلتزم موقع وصفة طبية بأعلى معايير الدقة العلمية والمهنية في إعداد محتواه الصحي. يخضع كل مقال لمراجعة طبية متخصصة وتدقيق علمي ولغوي دقيق قبل نشره.
مصادرنا مستقاة من أبرز المجلات الطبية المحكّمة والمنظمات الصحية الدولية المعتمدة، كمنظمة الصحة العالمية (WHO) والمعاهد الوطنية للصحة الأميركية (NIH) والاتحاد الدولي للثلاسيميا (TIF).
نحن لا نروّج لأي منتج أو علاج تجاري، ومحتوانا مستقل تماماً عن أي تمويل دوائي أو تجاري. هدفنا الوحيد هو تمكين القارئ من معلومة صحية موثوقة ومبنية على الدليل العلمي.
📋 البروتوكولات العلمية والإرشادات الطبية الرسمية المعتمدة
- دليل الاتحاد الدولي للثلاسيميا (TIF) — الإصدار الرابع 2021: الإرشادات الدولية الأشمل لإدارة الثلاسيميا المعتمدة على نقل الدم.
- إرشادات المعاهد الوطنية للصحة الأميركية (NIH/NHLBI) 2025: بروتوكولات التشخيص والمتابعة وعلاج تراكم الحديد في الثلاسيميا.
- موافقة إدارة الغذاء والدواء الأميركية (FDA) 2023: اعتماد أول علاجين جينيين للثلاسيميا (Casgevy® وZynteglo®).
- برنامج الفحص الصحي لما قبل الزواج — وزارة الصحة السعودية 2004 (محدَّث 2024): إلزامية فحص الثلاسيميا وفقر الدم المنجلي قبل عقد الزواج.
- دليل وزارة الصحة الإماراتية للأمراض الوراثية 2023: بروتوكولات الكشف المبكر والاستشارة الوراثية لحاملي الثلاسيميا.
- إرشادات الجمعية الأوروبية لأمراض الدم (EHA) 2024: توصيات علاج بيتا ثلاسيميا غير المعتمدة على نقل الدم وإدارة الثلاسيميا الوسطى.
المصادر والمراجع
- Taher, A. T., Weatherall, D. J., & Cappellini, M. D. (2018). Thalassaemia. The Lancet, 391(10116), 155-167. DOI: 10.1016/S0140-6736(17)31822-6
مراجعة شاملة لأنواع الثلاسيميا وتصنيفاتها وبروتوكولات العلاج الحديثة. - Musallam, K. M., et al. (2021). Iron overload in non-transfusion-dependent thalassemia: a clinical perspective. Blood Reviews, 46, 100745. DOI: 10.1016/j.blre.2020.100745
دراسة توضح آليات تراكم الحديد حتى في مرضى الثلاسيميا غير المعتمدين على نقل الدم. - Kattamis, A., et al. (2022). Changing patterns in the epidemiology of β-thalassemia. European Journal of Haematology, 108(2), 99-111. DOI: 10.1111/ejh.13710
دراسة وبائية عن تغير أنماط انتشار بيتا ثلاسيميا في أوروبا والشرق الأوسط. - Frangoul, H., et al. (2021). CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. New England Journal of Medicine, 384(3), 252-260. DOI: 10.1056/NEJMoa2031054
الدراسة المحورية التي أثبتت فعالية العلاج الجيني بتقنية كريسبر للثلاسيميا. - Locatelli, F., et al. (2022). Betibeglogene Autotemcel Gene Therapy for Non-β⁰/β⁰ Genotype β-Thalassemia. New England Journal of Medicine, 386(5), 415-427. DOI: 10.1056/NEJMoa2113206
دراسة العلاج الجيني Zynteglo® ونتائجه السريرية في تقليل الاعتماد على نقل الدم. - Kautz, L., et al. (2014). Identification of erythroferrone as an erythroid regulator of iron metabolism. Nature Genetics, 46(7), 678-684. DOI: 10.1038/ng.2996
الدراسة التي اكتشفت بروتين الإريثروفيرون ودوره في تثبيط الهيبسيدين. - World Health Organization (WHO). Sickle-cell disease and other haemoglobin disorders — Fact Sheet.
صحيفة وقائع منظمة الصحة العالمية عن اضطرابات الهيموغلوبين الوراثية. - Centers for Disease Control and Prevention (CDC). Thalassemia — CDC Information Page.
صفحة مراكز السيطرة على الأمراض والوقاية منها المخصصة للثلاسيميا. - National Heart, Lung, and Blood Institute (NHLBI) — NIH. Thalassemias — Health Topics.
مرجع شامل من المعاهد الوطنية للصحة الأميركية عن أنواع الثلاسيميا وتشخيصها وعلاجها. - U.S. Food and Drug Administration (FDA). FDA Approves First Gene Therapies for Sickle Cell Disease and Thalassemia (2023).
بيان FDA بالموافقة على العلاجات الجينية. - Thalassaemia International Federation (TIF). Guidelines for the Management of Transfusion-Dependent Thalassaemia, 4th Edition (2021).
الدليل الإرشادي الدولي الأهم لإدارة الثلاسيميا المعتمدة على نقل الدم. - Weatherall, D. J. (2010). The Thalassaemia Syndromes. 4th Edition. Wiley-Blackwell.
الكتاب المرجعي الأبرز في العالم عن متلازمات الثلاسيميا. - Hoffbrand, A. V., & Steensma, D. P. (2024). Hoffbrand’s Essential Haematology. 8th Edition. Wiley-Blackwell.
مرجع أكاديمي أساسي في أمراض الدم يشمل فصلاً تفصيلياً عن الثلاسيميا. - Cappellini, M. D., et al. (2020). Guidelines for the Management of Non-Transfusion-Dependent Thalassaemia. Thalassaemia International Federation.
دليل إدارة الثلاسيميا غير المعتمدة على نقل الدم. - Ware, R. E. (2020). Thalassemia: Current approaches in an evolving landscape. Scientific American — Health.
مقالة مبسّطة تشرح التطورات الحديثة في علاج الثلاسيميا للقارئ العام.
قراءات إضافية ومصادر للتوسع
- Piel, F. B., & Weatherall, D. J. (2014). The α-Thalassemias. New England Journal of Medicine, 371(20), 1908-1916. DOI: 10.1056/NEJMra1404415
لماذا نقترح عليك قراءته؟ هذه المراجعة تُعَدّ المرجع الأبرز لفهم ألفا ثلاسيميا تحديداً بتفصيل جزيئي وسريري ممتاز. - Rivella, S. (2012). The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood, 119(16), 3651-3656. DOI: 10.1182/blood-2011-12-396721
لماذا نقترح عليك قراءته؟ يشرح بعمق الآلية المركزية لتكوّن الدم غير الفعّال ودور الإريثروبويتين في حلقة المرض المفرغة. - Higgs, D. R., Engel, J. D., & Stamatoyannopoulos, G. (2012). Thalassaemia. The Lancet, 379(9813), 373-383. DOI: 10.1016/S0140-6736(11)60283-3
لماذا نقترح عليك قراءته؟ مراجعة شاملة من أبرز الباحثين في المجال تُغطي الأساس الجزيئي والسريري والعلاجي لجميع أنواع الثلاسيميا.
إذا وجدت في هذا المقال فائدة واحدة أضاءت لك جانباً لم تكن تعرفه، فشاركه مع من تحب — فقد يكون هناك حامل للسمة لا يعلم بذلك بعد، ومعلومة واحدة قد تُغيّر مسار حياة عائلة بأكملها. ولا تتردد في استشارة طبيب أمراض الدم أو الوراثة إذا كان لديك أي سؤال لم تجد إجابته هنا — صحتك وصحة أطفالك تستحق كل اهتمام.
⚠️ تحذير طبي وإخلاء مسؤولية — موقع وصفة طبية
المحتوى المقدَّم في هذا المقال هو للأغراض التثقيفية والتوعوية الصحية العامة حصراً، ولا يُشكّل بأي حال من الأحوال تشخيصاً طبياً أو وصفة علاجية أو بديلاً عن الاستشارة الطبية المتخصصة.
لا يتحمّل موقع وصفة طبية أي مسؤولية عن قرارات صحية يتّخذها القارئ بناءً على المعلومات الواردة هنا دون الرجوع إلى طبيب مختص. الجرعات والأدوية والمكملات المذكورة هي للإيضاح العلمي فقط وقد تختلف بحسب الحالة الفردية.
إذا كنت تعاني من أعراض أو لديك استفسار صحي، يُرجى مراجعة طبيبك المعالج أو أقرب مرفق صحي معتمد.
د. سوزان عبد الحميد السعدي — استشارية أمراض الدم وزرع الخلايا الجذعية