متلازمة دياموند بلاكفان (فقر الدم الماسي): أسباب فشل نخاع العظم، الأعراض، وخيارات العلاج المتاحة
ما الذي يجعل هذا المرض النادر مختلفاً عن أي فقر دم آخر؟

متلازمة دياموند بلاكفان (فقر الدم الماسي) هي اضطراب وراثي نادر يصيب نخاع العظم، فيعجز عن إنتاج خلايا الدم الحمراء (Red Blood Cells) بكميات كافية منذ الأشهر الأولى من العمر. تُقدَّر نسبة الإصابة بحالة واحدة تقريباً لكل 100,000 إلى 200,000 ولادة حية عالمياً. يُشخَّص غالبية المرضى قبل بلوغهم العام الأول، ويرتبط المرض بطفرات جينية تؤثر في بروتينات الريبوسومات المسؤولة عن بناء الخلايا.
د. فارس أحمد الرشيدي — طبيب مختص في الأمراض الوراثية والجينوم
د. سوزان عبد الحميد السعدي — استشارية أمراض الدم وزرع الخلايا الجذعية
د. عبد الرحمن الصباغ — خبير طب الأطفال وحديثي الولادة
المعلومات الواردة في هذا المقال هي للتثقيف الصحي فقط ولا تغني بأي حال عن استشارة الطبيب المختص.
- شحوب شديد مستمر عند الرضيع في الأشهر الأولى يستوجب تحليل دم فورياً (CBC + خلايا شبكية).
- خلايا شبكية أقل من 1% مع هيموغلوبين منخفض = إحالة عاجلة لاختصاصي دم الأطفال.
- افحص يدي طفلك — الإبهام ثلاثي السلاميات أو الوجه المثلثي الصغير قد تكون علامات مرافقة.
- لا تعطِ طفلك مكملات حديد قبل تشخيص دقيق — قد تضره بتراكم الحديد في أعضائه.
- احتفظ بملف طبي منظم: تحاليل، جرعات، مواعيد نقل الدم، ومنحنيات النمو.
- فحص الفيريتين كل 3 أشهر على الأقل إذا كان طفلك على نقل دم دوري.
- الطفل على الكورتيزون يحتاج كالسيوم + فيتامين D يومياً لحماية عظامه.
- راقب علامات الخطر: حرارة مفاجئة، شحوب شديد، ضيق تنفس — الطوارئ فوراً.
- المرض ليس ناتجاً عن نقص حديد — بل عن خلل في جينات بروتينات الريبوسوم يمنع النخاع من إنتاج كريات حمراء ناضجة.
- 40% إلى 45% من الحالات ناتجة عن طفرات تلقائية — غياب التاريخ العائلي لا ينفي التشخيص.
- زراعة النخاع من شقيق متطابق تحقق شفاءً كاملاً بنسبة تتجاوز 90% قبل سن العاشرة.
هل أخبرك طبيب طفلك يوماً أن شحوبه ليس مجرد نقص حديد عابر، بل قد يكون شيئاً أعمق من ذلك؟ إن كنت أباً أو أماً لاحظت أن رضيعك شاحب باستمرار، يرضع بصعوبة، ويبدو متعباً أكثر من أقرانه، فمن الطبيعي أن يتسلل إليك القلق. هذا المقال كُتب لك تحديداً؛ ليأخذ بيدك خطوة بخطوة في فهم ما يحدث داخل جسم طفلك، وليمنحك أدوات عملية للتعامل مع التشخيص والعلاج بثقة ووعي، دون تهويل ودون تهوين.
تخيّل هذا المشهد: أم سعاد، من الرياض، لاحظت أن رضيعتها ذات الأربعة أشهر “نورة” تبدو شاحبة على نحو غير طبيعي رغم رضاعتها الطبيعية. زارت طبيب الأطفال الذي طلب تحليل دم شاملاً (CBC)، فأظهرت النتائج انخفاضاً حاداً في الهيموغلوبين مع شبه غياب للخلايا الشبكية (Reticulocytes) — وهي الخلايا الحمراء الفتية التي يصنعها النخاع. أُحيلت نورة إلى اختصاصي أمراض دم الأطفال، وبعد خزعة نخاع العظم وفحص جيني، تأكد التشخيص: متلازمة دياموند بلاكفان. بدأت نورة العلاج بالكورتيزون واستجابت خلال أسابيع. الخلاصة العملية؟ لا تتجاهل الشحوب المستمر عند رضيعك، واطلب تحليل دم مبكراً؛ فالتشخيص المبكر يفتح أبواب العلاج الفعّال.
اقرأ أيضاً:
- فقر الدم (الأنيميا): الأسباب، الأعراض، وطرق العلاج الطبية والغذائية
- الفحوصات الطبية الدورية: متى يجب أن تبدأ وما الذي تحتاجه في كل عمر؟
ما هي متلازمة دياموند بلاكفان (Diamond-Blackfan Anemia) بالضبط؟
لنبدأ من الأساس: نخاع العظم (Bone Marrow) هو المصنع الذي ينتج خلايا دمك. تخيّله كمصنع سيارات ضخم يعمل على مدار الساعة لإنتاج ثلاثة أنواع من “المركبات”: خلايا الدم الحمراء التي تنقل الأكسجين، وخلايا الدم البيضاء التي تحارب العدوى، والصفائح الدموية التي توقف النزيف. في متلازمة دياموند بلاكفان، يتوقف خط إنتاج واحد فقط عن العمل بكفاءة — وهو خط خلايا الدم الحمراء — بينما يظل إنتاج البيضاء والصفائح طبيعياً في معظم الحالات. النتيجة؟ فقر دم شديد يظهر مبكراً جداً في حياة الطفل.
هذا التعريف يقودنا إلى سؤال جوهري كثيراً ما يطرحه الأهل: ما الفرق بين فقر الدم العادي وفقر الدم الناتج عن متلازمة دياموند بلاكفان؟ الفرق جذري. فقر الدم الشائع عند الأطفال — كفقر الدم بعوز الحديد (Iron Deficiency Anemia) — ينتج عن نقص مادة خام يحتاجها المصنع. أعطِ المصنع الحديد، ويعود للعمل. لكن في مرض Diamond-Blackfan Anemia، المشكلة ليست في المادة الخام؛ بل في الآلات نفسها. الخلايا الجذعية المسؤولة عن التحول إلى كريات حمراء ناضجة لا تستطيع إتمام عملها بسبب خلل جيني عميق في بنيتها الداخلية. لذلك، مكملات الحديد وحدها لن تحل المشكلة أبداً — بل قد تضر الطفل لاحقاً بتراكم الحديد في أعضائه.
حقيقة طبية: متلازمة دياموند بلاكفان هي واحدة من مجموعة نادرة تُسمى “متلازمات فشل نخاع العظم الموروثة” (Inherited Bone Marrow Failure Syndromes – IBMFS)، وتختلف تماماً عن فقر الدم الناتج عن سوء التغذية أو النزف المزمن.
اقرأ أيضاً: مرض الثلاسيميا: رحلة طبية مفصلة من الأعراض المبكرة إلى أحدث بروتوكولات العلاج
كيف اكتُشفت هذه المتلازمة ومن أعطاها اسمها؟
القصة بدأت عام 1938، حين وصف الطبيبان الأميركيان لويس دياموند (Louis Diamond) وكينيث بلاكفان (Kenneth Blackfan) مجموعة من الأطفال الرضع الذين يعانون فقر دم شديداً مع نخاع عظم يبدو طبيعياً تقريباً — باستثناء غياب شبه تام للخلايا السليفة الحمراء (Erythroid Precursors). كان هذا الوصف السريري الأول لما بات يُعرف لاحقاً باسم متلازمة دياموند بلاكفان. ومنذ ذلك الحين، تطورت معرفتنا بالمرض تطوراً هائلاً؛ إذ لم نعد نكتفي بالوصف السريري، بل أصبحنا نفهم الطفرات الجينية المسببة على المستوى الجزيئي.
ما يثير الاهتمام أن دياموند وبلاكفان عملا في مستشفى بوسطن للأطفال (Boston Children’s Hospital)، أحد أعرق مراكز طب الأطفال في العالم. ولولا ملاحظتهما الدقيقة لحالات بدت “غامضة” آنذاك، لربما تأخر اكتشاف هذا المرض عقوداً. هذا يذكرنا بأن الطب يتقدم حين يطرح الأطباء أسئلة جريئة أمام حالات لا تتبع القواعد المعتادة.
لماذا لا يصنع نخاع عظم طفلك كريات حمراء كافية؟
الطفرات الجينية: مفتاح اللغز
السبب الجذري لمتلازمة دياموند بلاكفان يكمن في طفرات جينية (Genetic Mutations) تصيب جينات مسؤولة عن بناء مكونات الريبوسوم (Ribosome). فما هو الريبوسوم؟ تخيّله كآلة مجهرية داخل كل خلية، وظيفتها قراءة التعليمات الجينية وتحويلها إلى بروتينات — تماماً كطابعة ثلاثية الأبعاد تأخذ ملفاً رقمياً وتصنع منه قطعة ملموسة. عندما تتعطل هذه “الطابعة” بسبب طفرة في أحد مكوناتها، لا تستطيع الخلية الجذعية إنتاج البروتينات اللازمة لتنضج وتصبح كرية حمراء سليمة.
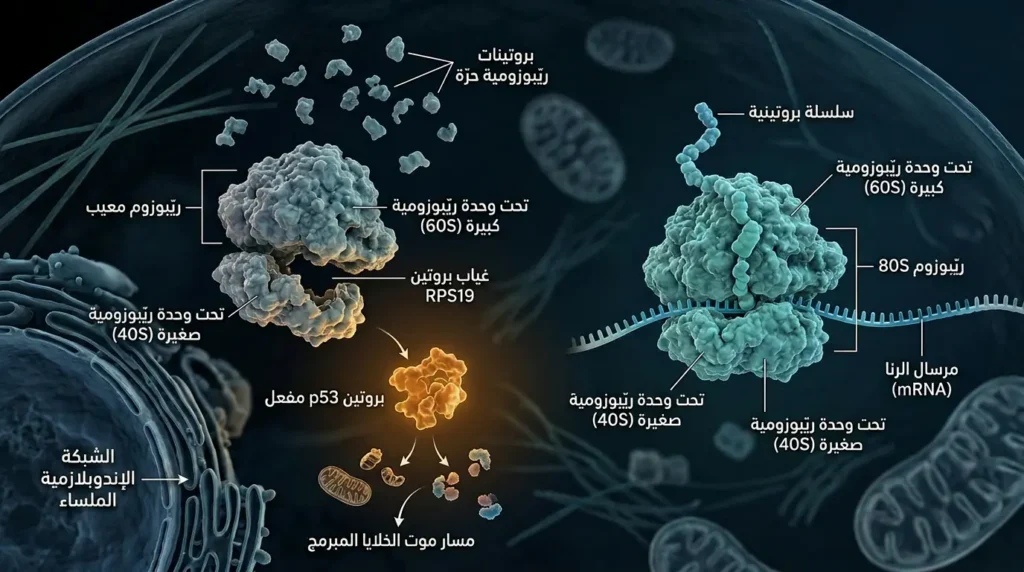
أشهر جين مسؤول هو RPS19، الذي يُشفِّر بروتيناً ريبوسومياً صغيراً (Ribosomal Protein S19). طفرات هذا الجين وحده تفسر نحو 25% من جميع حالات فقر الدم دياموند بلاكفان وفقاً لبيانات السجل الأميركي لمتلازمة دياموند بلاكفان (Diamond Blackfan Anemia Registry). لكنه ليس الجين الوحيد؛ فقد اكتُشفت حتى اليوم أكثر من 20 طفرة جينية مختلفة في جينات ريبوسومية أخرى مثل RPL5، RPL11، RPS26، RPL35A وغيرها، وكل منها يؤدي إلى الصورة السريرية ذاتها بدرجات متفاوتة.
كيف ينتقل المرض وراثياً؟
النمط الوراثي الأغلب هو الوراثة الجسمية السائدة (Autosomal Dominant)، ما يعني أن نسخة واحدة معيبة من الجين — موروثة من أحد الوالدين فقط — تكفي لإحداث المرض. لكن انتبه: ليس كل والد يحمل الطفرة يظهر عليه المرض بوضوح؛ فبعض الحاملين يعانون أعراضاً خفيفة جداً أو لا أعراض البتة — وهو ما يُعرف بالتعبير الجيني المتفاوت (Variable Expressivity). هذا يعني أن أباً أو أماً قد يحملان الطفرة دون أن يعرفا، ثم يُفاجَأان بطفل مصاب بصورة كاملة.
من ناحية أخرى، في 40% إلى 45% من الحالات تقريباً، تكون الطفرة تلقائية جديدة (De Novo Mutation) — أي أنها ظهرت لأول مرة في الطفل دون أن يرثها من أي من والديه. هل يمكن أن تحدث متلازمة دياموند بلاكفان بدون تاريخ عائلي؟ نعم، وهذا يحدث في نسبة كبيرة من الحالات. لذلك لا ينبغي أن يقول أحد: “مستحيل أن يكون طفلي مصاباً لأنه لا أحد في عائلتنا لديه هذا المرض.”
ومضة علمية: أثبتت دراسة نُشرت في مجلة Blood عام 2023 أن بعض المرضى الذين لم يُعثر لديهم على طفرة في الجينات الريبوسومية المعروفة يحملون طفرات في جينات أخرى تؤثر في مسارات مرتبطة ببروتين p53 — وهو “حارس الجينوم” الذي ينظّم موت الخلايا المبرمج (Apoptosis). هذا يفسر لماذا تموت الخلايا السليفة الحمراء قبل أن تنضج.
يقول الدكتور فارس أحمد الرشيدي — طبيب مختص في الأمراض الوراثية والجينوم في موقع وصفة طبية:
“الفحص الجيني لم يعد ترفاً بل ضرورة تشخيصية في حالات فقر الدم الوراثي النادر. تحديد الطفرة بدقة يساعدنا ليس فقط في تأكيد التشخيص، بل في تقدير المخاطر المستقبلية للطفل — بما فيها احتمال الإصابة بأورام معينة — وفي تقديم مشورة وراثية دقيقة للعائلة إذا رغبت في الإنجاب مجدداً.”
اقرأ أيضاً: ما هي تقنية كريسبر (CRISPR-Cas9) وكيف تعيد كتابة مستقبل الطب البشري؟
ما الفرق الحقيقي بين فقر الدم العادي وفقر الدم في هذه المتلازمة؟
هذه النقطة تستحق التوقف عندها لأنها المكان الذي يقع فيه كثير من الأهل في حيرة. حين يسمع الأب أو الأم كلمة “فقر دم”، يتبادر إلى الذهن فوراً: “سنعطيه حديداً وينتهي الأمر.” لكن الواقع أعقد من ذلك في حالة فقر الدم الوراثي النادر.
في فقر الدم بعوز الحديد، تكون مخازن الحديد فارغة — كخزان وقود سيارة يحتاج إلى تعبئة. تعبّئ الخزان (تعطي الحديد)، والمحرك (نخاع العظم) يعمل وينتج كريات حمراء طبيعية. أما في متلازمة دياموند بلاكفان، فالخزان ممتلئ بالوقود لكن المحرك نفسه معطّل. الحديد موجود، والفيتامينات موجودة، لكن خط إنتاج الكريات الحمراء في النخاع متوقف بسبب خلل في “البرمجة الداخلية” للخلايا الجذعية. هذا الفارق الجوهري يعني أن العلاج مختلف تماماً، والمتابعة أكثر تعقيداً، والأفق العلاجي يتطلب فريقاً طبياً متخصصاً.
ثمة فرق مخبري واضح أيضاً: في فقر الدم بعوز الحديد، تجد الخلايا الشبكية مرتفعة (لأن النخاع “يحاول” التعويض)، بينما في متلازمة دياموند بلاكفان تكون الخلايا الشبكية منخفضة جداً أو شبه غائبة — وهذا مؤشر تشخيصي حاسم.
| وجه المقارنة | فقر الدم بعوز الحديد | متلازمة دياموند بلاكفان |
|---|---|---|
| السبب الجذري | نقص مادة خام (الحديد) يحتاجها النخاع | خلل جيني في بروتينات الريبوسوم — آلة النخاع معطلة |
| عمر الظهور | غالباً بعد 6 أشهر مع تنوع الغذاء | الأشهر الأولى من العمر — أحياناً منذ الولادة |
| الخلايا الشبكية | طبيعية أو مرتفعة — النخاع يحاول التعويض | منخفضة جداً أو شبه غائبة — النخاع لا ينتج |
| حجم الكريات الحمراء (MCV) | صغير (Microcytic) | كبير (Macrocytic) |
| مستوى الحديد / الفيريتين | منخفض | طبيعي أو مرتفع (مع نقل الدم) |
| استجابة لمكملات الحديد | ممتازة — يتحسن الهيموغلوبين في أسابيع | لا استجابة — بل قد يضر بتراكم الحديد |
| الكريات البيضاء والصفائح | طبيعية | طبيعية في معظم الحالات |
| التشوهات الخلقية المرافقة | لا | نعم — في نحو 50% من المرضى |
| الوراثة | لا علاقة للوراثة — مكتسب | وراثي (سائد) أو طفرة تلقائية جديدة |
| العلاج الجذري | مكملات حديد — نتائج ممتازة | كورتيزون / نقل دم / زراعة نخاع |
| الخطورة المستقبلية | منخفضة — يُشفى تماماً مع التغذية الصحيحة | مزمن — يتطلب متابعة مدى الحياة مع خطر أورام محدود |
نقطة تستحق الانتباه: إذا أظهر تحليل دم طفلك الرضيع فقر دم مع خلايا شبكية منخفضة جداً (أقل من 1%)، فهذا ليس فقر دم غذائي عادي. اطلب من طبيب طفلك تحويله فوراً إلى اختصاصي أمراض دم الأطفال.
ما الأعراض والعلامات التي تكشف عن المتلازمة مبكراً؟
علامات نقص الكريات الحمراء
معظم الأطفال المصابين بمتلازمة دياموند بلاكفان تظهر عليهم الأعراض خلال الأشهر الثلاثة إلى الستة الأولى من العمر. الشحوب الشديد هو العلامة الأبرز — وليس المقصود هنا شحوباً خفيفاً يلاحظه الأهل أحياناً، بل شحوباً واضحاً في الوجه والشفتين وراحة اليدين، يُلفت انتباه حتى الغرباء. يرافق ذلك إرهاق ملحوظ عند الرضيع: ينام أكثر من المعتاد، يرضع ببطء وبضعف، ويبكي بصوت خافت. سرعة ضربات القلب (Tachycardia) علامة أخرى مهمة؛ إذ يحاول قلب الطفل الصغير ضخ الدم بسرعة أكبر لتعويض نقص الأكسجين الناتج عن قلة الكريات الحمراء.
فما الذي يجب أن تفعله الآن إذا لاحظت هذه العلامات؟ لا تنتظر الزيارة الدورية. خذ طفلك إلى طبيب الأطفال اليوم، واطلب تحليل صورة دم كاملة (CBC) مع عدد الخلايا الشبكية. هذا التحليل البسيط قد يكشف الكثير.
اقرأ أيضاً: رفض الرضيع للثدي: الأسباب الخفية والحلول العملية لإنقاذ رحلة الرضاعة
التشوهات الجسدية المرافقة: أكثر من مجرد فقر دم
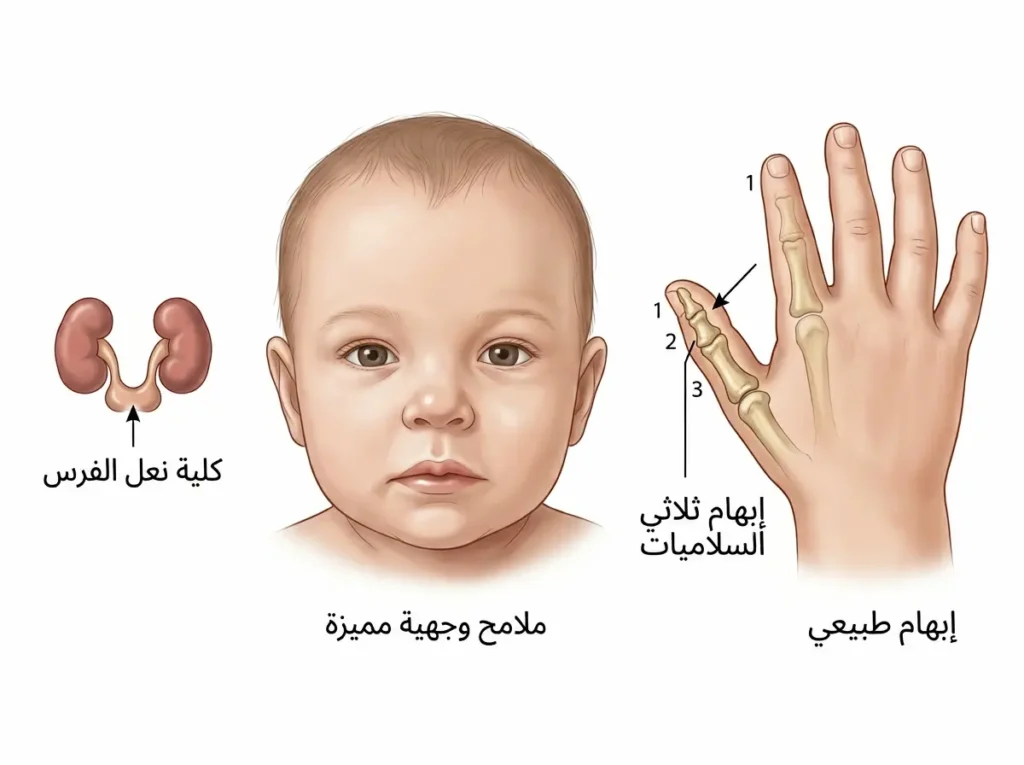
ما يميز أعراض متلازمة دياموند بلاكفان عن أنواع فقر الدم الأخرى هو أن نحو 50% من المرضى يولدون بتشوهات خلقية جسدية مرافقة. هذه التشوهات ليست صدفة؛ بل تعكس الدور الحيوي للريبوسومات في نمو الأنسجة المختلفة خلال مرحلة تكوّن الجنين.
من أبرز هذه التشوهات:
- تشوهات الإبهام والأصابع: قد يولد الطفل بإبهام ثلاثي السلاميات (Triphalangeal Thumb) بدلاً من اثنتين، أو بإبهام مسطح، أو — نادراً — بغياب الإبهام تماماً. افحص يدي طفلك بعناية.
- تشوهات الوجه والرأس: وجه صغير مثلثي الشكل، أنف أفطس، فك سفلي صغير (Micrognathia)، شفة علوية سميكة، وعينان متباعدتان (Hypertelorism). هذه الملامح قد تكون خفيفة ولا تُلاحَظ إلا بعين مدربة.
- عيوب القلب الخلقية: مثل عيب الحاجز البطيني (Ventricular Septal Defect – VSD)، وتُكتشف عادة بالإيكو القلبي (Echocardiography).
- تشوهات الكلى والمسالك البولية: كلية حذوية (Horseshoe Kidney) أو غياب إحدى الكليتين.
- قصر القامة وتأخر النمو: يلاحظ كثير من الأهل أن طفلهم أقصر من أقرانه في العمر نفسه. هذا ليس بالضرورة دليلاً على سوء تغذية؛ بل قد يكون جزءاً من الصورة السريرية للمتلازمة.
معلومة سريعة: ليس كل طفل مصاب بمتلازمة دياموند بلاكفان يعاني تشوهات جسدية. نحو نصف المرضى لا يظهر عليهم أي تشوه مرئي، ما يجعل التشخيص يعتمد أساساً على تحاليل الدم وفحص النخاع.
المختبر الفسيولوجي — للمهتمين بالتفاصيل العلمية الدقيقة
ما يحدث داخل الخلية المصابة بمتلازمة دياموند بلاكفان أشبه بسلسلة دومينو جزيئية تبدأ بخطأ واحد وتنتهي بانهيار خط إنتاج بأكمله. لنغُص في التفاصيل.
الريبوسوم البشري مكوّن من وحدتين فرعيتين: الكبرى (60S) والصغرى (40S)، وكل منهما يضم عشرات البروتينات الريبوسومية إلى جانب الحمض النووي الريبوسومي (rRNA). حين تحدث طفرة في جين مثل RPS19 (الذي يشفّر بروتيناً في الوحدة الفرعية 40S)، يختل تجميع الريبوسوم (Ribosome Assembly). النتيجة الأولى هي ما يُعرف بـ “الإجهاد الريبوسومي” (Ribosomal Stress أو Nucleolar Stress). هذا الإجهاد يُطلق إشارة تنبيه داخل الخلية عبر تفعيل مسار بروتين p53 — وهو بروتين ورمي كابت (Tumor Suppressor) يلعب دور “الحارس” الذي يقرر مصير الخلية: هل تتوقف عن الانقسام أم تدخل في الموت المبرمج (Apoptosis)؟
في الخلايا السليفة الحمراء (Erythroid Progenitors)، يكون تفعيل p53 كارثياً. هذه الخلايا حساسة بشكل استثنائي لمستويات p53 المرتفعة — أكثر من أي نوع خلوي آخر في الجسم. لماذا؟ لأن عملية تكوّن الكريات الحمراء (Erythropoiesis) تتطلب معدلات انقسام سريعة للغاية وترجمة بروتينية (Protein Translation) مكثفة لإنتاج كميات هائلة من الهيموغلوبين. أي خلل في قدرة الريبوسوم على الترجمة يعني أن الخلية لا تستطيع مواكبة هذا الطلب الإنتاجي العالي، فيتفعل p53 ويأمر الخلية بالانتحار قبل أن تنضج.
هذا يفسر لماذا يتأثر خط الكريات الحمراء تحديداً بينما تبقى خطوط الكريات البيضاء والصفائح سليمة نسبياً: فهذه الأخيرة أقل اعتماداً على الترجمة البروتينية المكثفة وأقل حساسية لتفعيل p53. كما أن بعض الدراسات الحديثة (2024) تشير إلى أن اضطراب معالجة rRNA في النُّوَيَّة (Nucleolus) يؤدي إلى تراكم بروتينات ريبوسومية حرة غير مُجمَّعة (Free Ribosomal Proteins)، وهذه البروتينات الحرة — خصوصاً RPL5 وRPL11 — ترتبط مباشرة ببروتين MDM2 (E3 Ubiquitin Ligase)، مما يمنعه من تحطيم p53. والنتيجة: ارتفاع مستمر في p53 وموت خلوي مبرمج متواصل.
الجدير بالذكر أن هذا الفهم الجزيئي العميق فتح آفاقاً علاجية جديدة: إذا استطعنا تثبيط p53 جزئياً أو تعزيز تجميع الريبوسومات بطرق دوائية أو جينية، فقد نتمكن مستقبلاً من إعادة خط إنتاج الكريات الحمراء إلى العمل.
رقم لافت: أظهرت بيانات السجل الفرنسي لمتلازمة دياموند بلاكفان أن خطر الإصابة بأورام خبيثة — خصوصاً متلازمة خلل التنسج النخاعي (MDS) وابيضاض الدم النقوي الحاد (AML) — يرتفع لدى مرضى هذه المتلازمة بنسبة تصل إلى 5 أضعاف مقارنة بالسكان الطبيعيين. وهذا مرتبط بالخلل المزمن في مسار p53 وعدم استقرار الجينوم.
كيف يُشخَّص المرض؟ رحلة التشخيص الطبي خطوة بخطوة
الفحص السريري وملاحظة العلامات الأولية
يبدأ التشخيص عادة حين يلاحظ طبيب الأطفال أو الأهل شحوباً شديداً ومستمراً عند رضيع صغير. الطبيب المتمرس سيبحث فوراً عن علامات إضافية: هل هناك تشوه في الإبهام؟ هل ملامح الوجه غير نمطية؟ هل وزن الطفل وطوله أقل من المنحنى الطبيعي؟ كل ملاحظة — مهما بدت صغيرة — قد تكون قطعة من أحجية التشخيص.
التحاليل المخبرية: الخطوة الحاسمة
أول فحص يُطلب هو تعداد الدم الكامل (Complete Blood Count – CBC). في حالة متلازمة دياموند بلاكفان النموذجية، ستجد: هيموغلوبين منخفض (غالباً أقل من 7 غ/دل)، حجم الكريات الحمراء كبير (Macrocytic Anemia)، خلايا بيضاء وصفائح طبيعية. العلامة الأكثر دلالة هي انخفاض عدد الخلايا الشبكية (Reticulocyte Count) إلى مستويات متدنية جداً — أحياناً أقل من 0.5% — ما يدل على أن النخاع لا يصنع كريات حمراء جديدة.
فحص آخر مهم هو مستوى الأدينوزين ديأميناز الكريات الحمراء (Erythrocyte Adenosine Deaminase – eADA)، الذي يكون مرتفعاً في نحو 80% إلى 85% من مرضى فقر الدم دياموند بلاكفان. كما يُلاحظ ارتفاع الهيموغلوبين الجنيني (Fetal Hemoglobin – HbF) عن المستويات الطبيعية لعمر الطفل.
| الفحص المخبري | القيمة الطبيعية (رضيع) | النتيجة في متلازمة دياموند بلاكفان | الدلالة التشخيصية |
|---|---|---|---|
| الهيموغلوبين (Hgb) | 10 – 14 غ/دل | أقل من 7 غ/دل عادةً | فقر دم شديد — مؤشر رئيسي للتشخيص |
| الخلايا الشبكية (Reticulocytes) | 1% – 2% | أقل من 0.5% — شبه غائبة | العلامة التشخيصية الأكثر دلالة — النخاع لا ينتج كريات حمراء |
| حجم الكرية الحمراء (MCV) | 70 – 90 فمتوليتر | مرتفع — فوق 95 فمتوليتر | فقر دم كبير الكريات (Macrocytic Anemia) |
| الكريات البيضاء (WBC) | 6,000 – 17,000 /ميكروليتر | طبيعية في معظم الحالات | يُفرّق المتلازمة عن فقر الدم اللاتنسجي الشامل |
| الصفائح الدموية (Platelets) | 150,000 – 400,000 /ميكروليتر | طبيعية في معظم الحالات | خط الإنتاج الصفيحي محفوظ — يؤكد انتقائية الخلل |
| أدينوزين ديأميناز الكريات الحمراء (eADA) | منخفض أو طبيعي | مرتفع في 80% – 85% من الحالات | مؤشر بيوكيميائي داعم للتشخيص |
| الهيموغلوبين الجنيني (HbF) | يتناقص تدريجياً بعد الولادة | مرتفع بشكل غير متناسب مع العمر | علامة داعمة — يعكس التأخر في نضج خلايا الدم الحمراء |
| الفيريتين (Ferritin) | أقل من 200 نانوغرام/مل | مرتفع جداً مع نقل الدم المزمن (أكثر من 1,000) | يرصد تراكم الحديد — يستوجب العلاج الخالب فور الارتفاع |
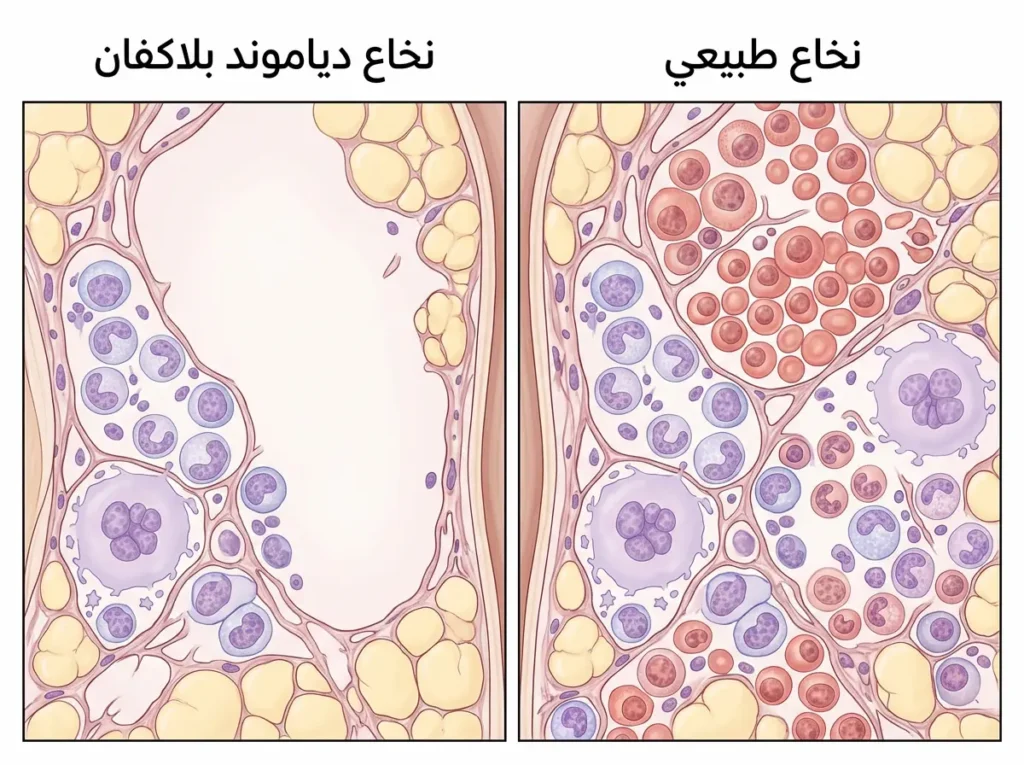
خزعة نخاع العظم: لماذا هي ضرورية؟
خزعة نخاع العظم (Bone Marrow Biopsy) ليست فحصاً سهلاً على الأهل نفسياً، لكنها حاسمة. في متلازمة دياموند بلاكفان، يُظهر النخاع خلوية طبيعية أو شبه طبيعية في جميع الخطوط — ما عدا خط الكريات الحمراء الذي يكون ناقصاً على نحو واضح (Erythroid Hypoplasia). تخيّل أنك تدخل مصنعاً فيه ثلاثة خطوط إنتاج: خطان يعملان بكامل طاقتهما، والثالث شبه خالٍ من العمال. هذا ما يراه الطبيب تحت المجهر.
الفحص الجيني: تأكيد التشخيص في العصر الحديث
بعد أن تتوافق الصورة السريرية والمخبرية، يأتي دور الفحص الجيني (Genetic Testing). يُجرى عادة باستخدام تقنيات التسلسل الجيني من الجيل التالي (Next-Generation Sequencing – NGS) التي تفحص عشرات الجينات الريبوسومية دفعة واحدة. العثور على طفرة مُسبِّبة يؤكد التشخيص نهائياً، لكن عدم العثور عليها لا ينفيه — لأن 30% إلى 35% من المرضى لا تُكتشف لديهم طفرة معروفة بالتقنيات المتاحة حالياً.
يقول الدكتور عبد الرحمن الصباغ — خبير طب الأطفال وحديثي الولادة في موقع وصفة طبية:
“حين يأتيني رضيع شاحب مع هيموغلوبين منخفض وخلايا شبكية شبه معدومة، أفكر فوراً في متلازمة دياموند بلاكفان حتى يثبت العكس. التشخيص المبكر في الأشهر الأولى يتيح لنا بدء العلاج في الوقت الأمثل ويحمي الطفل من مضاعفات فقر الدم الشديد على القلب والدماغ.”
اقرأ أيضاً: اليرقان الوليدي (صفار المواليد): الأسباب، درجات الخطورة، وخطوات العلاج الطبية
هل يمكن اكتشاف متلازمة دياموند بلاكفان قبل ولادة الطفل (في أثناء الحمل)؟
كثيراً ما يطرح الآباء الذين شُخِّص طفلهم الأول بهذا المرض سؤالاً مليئاً بالقلق: “هل سيتكرر الأمر في حملي القادم؟ وهل يمكننا معرفة ذلك مبكراً؟”
الإجابة تعتمد على التاريخ العائلي ومعرفة الطفرة الجينية. إذا كانت الطفرة الجينية المسببة للمرض لدى الطفل الأول أو أحد الوالدين معروفة ومحددة، فإن التشخيص الجيني قبل الولادة (Prenatal Genetic Diagnosis) يصبح ممكناً ودقيقاً للغاية. يُجرى ذلك عادة عبر أخذ خزعة من الزغابات المشيمية (CVS) في الأسابيع (10-12) من الحمل، أو فحص السائل الأمينوسي (Amniocentesis) في الأسابيع (15-18).
أما إذا لم يكن هناك تاريخ عائلي معروف، فقد يشك طبيب التوليد في وجود مشكلة خلال أجهزة الموجات فوق الصوتية (Ultrasound) الروتينية إذا لاحظ:
- تأخراً شديداً في نمو الجنين (IUGR).
- تشوهات جسدية كغياب الإبهام أو عيوب القلب.
- الاستسقاء الجنيني (Hydrops Fetalis)، وهو تورم وتراكم للسوائل في جسم الجنين يحدث عندما يعاني فقر دم حاداً داخل الرحم.
رسالة طمأنينة: إذا كنتِ تخططين لحمل جديد ولديكِ طفل مصاب بمتلازمة دياموند بلاكفان، فمن الضروري جداً زيارة عيادة الاستشارة الوراثية (Genetic Counseling) قبل الحمل. التقنيات الحديثة مثل الفحص الجيني للأجنة قبل الانغراس (PGD) خلال عمليات التلقيح الصناعي (IVF) تتيح الآن اختيار أجنة لا تحمل الطفرة الجينية المسببة للمرض.
كيف يُعالَج فقر الدم دياموند بلاكفان؟ الخيارات العلاجية بالتفصيل
⚠️ تحذير مهم: لا تبدأ أو توقف أي علاج لطفلك دون إشراف مباشر من اختصاصي أمراض دم الأطفال. المعلومات التالية تثقيفية وليست بديلاً عن الاستشارة الطبية الفردية.
العلاج بالكورتيكوستيرويدات (الكورتيزون): الخيار الأول
الكورتيكوستيرويدات — وتحديداً البريدنيزولون (Prednisolone) أو البريدنيزون (Prednisone) — هي الخط الأول لعلاج فقر الدم دياموند بلاكفان بالكورتيزون. آلية عملها ليست مفهومة بالكامل، لكن يُعتقد أنها تثبّط جزئياً مسار p53 وتعزز بقاء الخلايا السليفة الحمراء.
الجرعة الابتدائية عند الأطفال (بعد عمر 12 شهراً عادةً):
- تبدأ بجرعة 2 ملغ/كغ/يوم من البريدنيزولون مقسمة على جرعتين أو ثلاث.
- لا يُنصح عادة ببدء الكورتيزون قبل عمر السنة لتجنب تأثيراته الشديدة على نمو الرضيع.
- يُقيَّم الاستجابة خلال 2 إلى 4 أسابيع بمتابعة مستوى الهيموغلوبين والخلايا الشبكية.
عند الاستجابة:
- يُخفَّض الكورتيزون تدريجياً على مدى أسابيع إلى أشهر حتى الوصول إلى أقل جرعة فعالة — والهدف المثالي هو أقل من 0.5 ملغ/كغ/يوم، ويُفضَّل أن تكون أقل من 0.3 ملغ/كغ كل يومين.
- نسبة الاستجابة الأولية تتراوح بين 60% و80% من المرضى.
الآثار الجانبية — وهي كثيرة ويجب أن يعرفها كل أب وأم:
- تباطؤ النمو الطولي: أخطر أثر جانبي عند الأطفال. المتابعة الدورية لمنحنى النمو ضرورية.
- زيادة الوزن وتغير توزيع الدهون: الوجه القمري (Moon Face).
- هشاشة العظام: خصوصاً مع الاستخدام المطوّل.
- ارتفاع ضغط الدم وسكر الدم.
- ضعف المناعة وزيادة خطر العدوى.
- تقلبات مزاجية واضطرابات نوم.
فرط الجرعة: جرعات عالية فجائية قد تسبب ارتفاعاً حاداً في ضغط الدم، ارتفاع سكر الدم، واضطرابات نفسية حادة. لا تُعدّل الجرعة أبداً من تلقاء نفسك.
ماذا تفعل الآن؟ إذا كان طفلك على الكورتيزون، التزم بالجرعة المحددة بدقة، واحتفظ بسجل يومي للوزن والطول، وأخبر الطبيب فوراً إذا لاحظت علامات عدوى أو تغيراً سلوكياً غير طبيعي.
اقرأ أيضاً: مضادات الالتهاب: أنواعها، استخداماتها، والآثار الجانبية، الدليل الطبي الشامل
نقل الدم الدوري: شريان الحياة المؤقت
الأطفال الذين لا يستجيبون للكورتيزون — أو الذين تقل أعمارهم عن سنة — يحتاجون إلى نقل دم دوري (Chronic Red Blood Cell Transfusions). يُنقل الدم عادة كل 3 إلى 6 أسابيع للحفاظ على مستوى هيموغلوبين لا يقل عن 8 غ/دل.
نقل الدم يحسّن حالة الطفل على نحو ملحوظ ومباشر: يعود اللون إلى وجهه، يرضع أو يأكل بنشاط أكبر، ويصبح أكثر حيوية. لكن هذا التحسن يأتي بثمن خفي يتراكم مع الوقت.
الخطر الخفي: تراكم الحديد (Iron Overload)
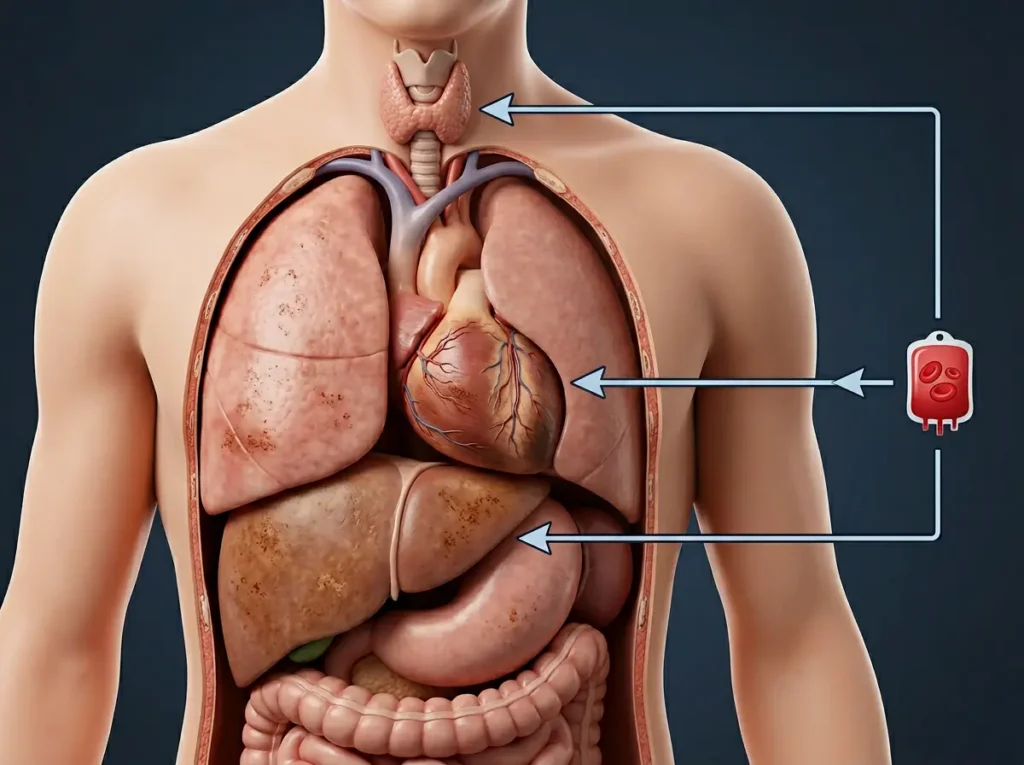
كل وحدة دم تُنقل إلى طفلك تحمل معها نحو 200 إلى 250 ملغ من الحديد. الجسم البشري لا يملك آلية طبيعية فعالة للتخلص من الحديد الزائد. وعليه فإن مضاعفات نقل الدم في متلازمة دياموند بلاكفان تتمحور على نحو رئيس حول تراكم الحديد في الأعضاء الحيوية — خصوصاً القلب والكبد والغدد الصماء — مسبباً أضراراً قد تكون خطيرة ودائمة: اعتلال عضلة القلب (Cardiomyopathy)، تليف الكبد، قصور الغدة الدرقية، السكري، وتأخر البلوغ.
📌 صندوق اقتباس طبي:
وفقاً لتقرير صادر عن المعاهد الوطنية للصحة (NIH) في الولايات المتحدة، فإن تراكم الحديد الناتج عن نقل الدم المزمن يُعَدُّ السبب الأول للوفاة المبكرة لدى المرضى الذين يعتمدون على نقل الدم طويل الأمد في أمراض فشل نخاع العظم عند الأطفال. المتابعة المنتظمة لمستوى الفيريتين (Serum Ferritin) والتصوير بالرنين المغناطيسي للقلب والكبد (MRI T2*) ضرورية لرصد التراكم مبكراً.
اقرأ أيضاً: قصور القلب (ضعف عضلة القلب): الأسباب، العلامات التحذيرية، وخيارات العلاج المتقدمة
العلاج الخالب للحديد (Iron Chelation Therapy): كيف نتخلص من الحديد الزائد؟
⚠️ تحذير دوائي: لا تبدأ أو تعدّل جرعة أي دواء خالب للحديد دون إشراف طبيب متخصص. الجرعات الخاطئة قد تسبب أضراراً كلوية أو سمعية أو بصرية خطيرة.
الأدوية الخالبة للحديد تعمل كـ “مغناطيس كيميائي” يلتقط جزيئات الحديد الحرة من الدم والأنسجة ويطرحها عبر البول أو البراز. أبرز الأدوية المستخدمة:
1. ديفيراسيروكس (Deferasirox – الاسم التجاري: Exjade أو Jadenu):
- الشكل: أقراص أو حبيبات تُذاب في الماء أو العصير.
- الجرعة عند الأطفال (من عمر سنتين فأكبر): تبدأ بـ 20 ملغ/كغ/يوم (لمستحضر Exjade) أو 14 ملغ/كغ/يوم (لمستحضر Jadenu) مرة واحدة يومياً. تُعدَّل الجرعة كل 3 إلى 6 أشهر بناءً على مستوى الفيريتين.
- الحد الأقصى: 40 ملغ/كغ/يوم (Exjade) أو 28 ملغ/كغ/يوم (Jadenu).
- الآثار الجانبية: اضطرابات هضمية (غثيان، إسهال)، ارتفاع كرياتينين الدم (يستوجب مراقبة وظائف الكلى شهرياً)، طفح جلدي، واضطرابات سمعية أو بصرية نادرة.
- فرط الجرعة: قد يسبب فشلاً كلوياً حاداً أو فشلاً كبدياً. في حال تناول جرعة مفرطة، توجّه إلى الطوارئ فوراً.
2. ديفيروكسامين (Deferoxamine – الاسم التجاري: Desferal):
- الشكل: حقن تحت الجلد أو وريدية.
- الجرعة عند الأطفال (من عمر 3 سنوات فأكبر): 20 إلى 40 ملغ/كغ/يوم تُعطى بالتسريب تحت الجلد على مدى 8 إلى 12 ساعة (عادة ليلاً عبر مضخة)، 5 إلى 7 أيام أسبوعياً.
- الآثار الجانبية: ألم وتورم مكان الحقن، اضطرابات سمعية وبصرية (يجب إجراء فحوصات سمع وبصر دورية)، تباطؤ النمو عند الجرعات العالية.
- فرط الجرعة: جرعات مفرطة قد تسبب انخفاض ضغط حاد، صدمة، واضطرابات بصرية دائمة.
3. ديفيريبرون (Deferiprone – الاسم التجاري: Ferriprox):
- يُستخدم أحياناً بالمشاركة مع ديفيراسيروكس أو ديفيروكسامين في حالات التراكم الشديد.
- الجرعة عند الأطفال (فوق 3 سنوات): 75 إلى 100 ملغ/كغ/يوم مقسمة على 3 جرعات.
- أخطر أثر جانبي: ندرة المحببات (Agranulocytosis) — انخفاض حاد ومفاجئ في كريات الدم البيضاء يجعل الطفل عرضة لعدوى مهددة للحياة. يستوجب فحص CBC أسبوعياً.
| الدواء | الشكل الدوائي | الجرعة (أطفال) | طريق الإعطاء | أخطر أثر جانبي | المتابعة المطلوبة |
|---|---|---|---|---|---|
| ديفيراسيروكس (Deferasirox) | أقراص / حبيبات تُذاب | 20 – 40 ملغ/كغ/يوم (Exjade) 14 – 28 ملغ/كغ/يوم (Jadenu) |
فموي — مرة يومياً | خطورة كلوية وكبدية | وظائف الكلى والكبد شهرياً — سمع وبصر سنوياً |
| ديفيروكسامين (Deferoxamine) | محلول للحقن | 20 – 40 ملغ/كغ/يوم | حقن تحت الجلد — 8 إلى 12 ساعة ليلاً | اضطرابات سمع وبصر دائمة | سمع وبصر كل 6 أشهر — نمو سنوياً |
| ديفيريبرون (Deferiprone) | أقراص / محلول فموي | 75 – 100 ملغ/كغ/يوم مقسمة على 3 جرعات | فموي — 3 مرات يومياً | ندرة المحببات (Agranulocytosis) — خطر عدوى قاتلة | CBC أسبوعياً بدون استثناء |
يقول المستشار الدوائي جاسم محمد مراد — خبير الصحة والإمداد الطبي في موقع وصفة طبية:
“أدوية خلب الحديد ليست مكملات غذائية يمكن تناولها بحرية. كل دواء منها يحتاج إلى مراقبة دقيقة لوظائف الكلى والكبد والسمع والبصر. أنصح الأهل بالاحتفاظ بتقويم واضح لمواعيد التحاليل الدورية ومواعيد الجرعات، وعدم تفويت أي موعد حتى لو بدا الطفل بصحة جيدة.”
زراعة الخلايا الجذعية: هل هي الحل الجذري النهائي؟
زراعة الخلايا الجذعية المكوِّنة للدم (Hematopoietic Stem Cell Transplantation – HSCT) هي العلاج الوحيد القادر على شفاء المتلازمة نهائياً. الفكرة بسيطة من حيث المبدأ: استبدال نخاع العظم المعيب بنخاع سليم من متبرع متوافق. لكن التنفيذ معقد والمخاطر حقيقية.
متى تكون الزراعة هي الخيار الأنسب؟
- عندما لا يستجيب الطفل للكورتيزون أو يعتمد على جرعات عالية منه.
- عندما يحتاج الطفل إلى نقل دم متكرر مع تراكم حديد متصاعد رغم العلاج الخالب.
- عندما يتوفر متبرع شقيق متطابق في نظام HLA (Human Leukocyte Antigen).
نسبة نجاح الزراعة:
وفقاً لبيانات نُشرت في مجلة Blood (2021)، فإن نسبة الشفاء من متلازمة دياموند بلاكفان بعد زراعة نخاع من شقيق متطابق تتجاوز 90% عند الأطفال الذين أُجريت لهم الزراعة قبل سن العاشرة. تنخفض هذه النسبة مع التقدم في العمر أو مع استخدام متبرعين غير أقارب.
المخاطر:
- مرض الطُّعم ضد الثوي (Graft-versus-Host Disease – GvHD).
- فشل الطُّعم (Graft Failure).
- عدوى خطيرة في فترة ضعف المناعة بعد الزراعة.
- مضاعفات طويلة الأمد على الخصوبة والنمو.
من المثير أن تعرف: في المملكة العربية السعودية، يوجد عدد من المراكز المتخصصة في زراعة الخلايا الجذعية للأطفال، منها مستشفى الملك فيصل التخصصي ومركز الأبحاث بالرياض، الذي يُجري عشرات عمليات الزراعة سنوياً لأمراض الدم الوراثية. استشر طبيب طفلك عن إمكانية التحويل إلى أحد هذه المراكز.
اقرأ أيضاً: كيف يعمل العلاج الكيميائي، آثاره الجانبية، وطرق التغلب عليها، الرحلة نحو التعافي
ما بعد الطفولة: تحديات البلوغ، الخصوبة، والانتقال لرعاية البالغين
معظم المقالات الطبية تتحدث عن متلازمة دياموند بلاكفان كمرض يُصيب الرضع والأطفال، لكن ماذا يحدث عندما يكبر هؤلاء الأطفال؟ بفضل التقدم الطبي المذهل، يصل معظم المرضى اليوم إلى مرحلة البلوغ والرشد ويعيشون حياة منتجة. لكن هذه المرحلة تجلب معها تحديات طبية من نوع آخر.
تأثير تراكم الحديد على الخصوبة والهرمونات
الأطفال الذين اعتمدوا على نقل الدم لسنوات طويلة قد يواجهون تأثيرات صامتة لتراكم الحديد في الغدة النخامية والخصيتين أو المبيضين. هذا التراكم قد يؤدي إلى حالة تُسمى “قصور الغدد التناسلية” (Hypogonadism)، ما يسبب تأخراً في البلوغ الطبيعي، أو تحديات في الخصوبة والإنجاب مستقبلاً. لذا، فإن الالتزام الصارم بالعلاج الخالب للحديد في الطفولة هو حرفياً استثمار في خصوبة طفلك ومستقبله العائلي.
الحمل لدى مريضات دياموند بلاكفان
هل يمكن لفتاة مصابة بالمتلازمة أن تحمل وتنجب مستقبلاً؟ نعم، هذا ممكن تماماً. لكن الحمل يُصنّف كحمل “عالي الخطورة” (High-Risk Pregnancy). فقر الدم قد يتفاقم بشدة خلال أشهر الحمل بسبب زيادة حجم بلازما الدم، ما قد يستدعي العودة لنقل الدم حتى لو كانت المريضة في حالة هدأة (Remission). كما تتطلب المريضة الحامل إشرافاً دقيقاً من طبيب أمراض دم وطبيب توليد متخصص.
مرحلة الانتقال (Transition of Care)
من أصعب المحطات هي انتقال المريض في سن الـ 18 من طبيب أطفال اعتاد عليه لسنوات، إلى طبيب أمراض دم للبالغين. ننصح الأهل بالبدء في تثقيف المريض اليافع حول مرضه وجرعات أدويته منذ سن الـ 14، ليكون مستعداً لتحمل مسؤولية صحته بثقة عندما يكبر.
ما المستجدات البحثية والعلاجات الجينية قيد التطوير؟
المجتمع العلمي لا يقف مكتوف الأيدي أمام هذا المرض النادر. هناك عدة محاور بحثية واعدة:
العلاج الجيني (Gene Therapy): تجارب مبكرة تستخدم ناقلات فيروسية (Viral Vectors) لإدخال نسخة سليمة من الجين المعيب إلى الخلايا الجذعية للمريض نفسه، ثم إعادتها إلى جسمه. نُشرت نتائج أولية مشجعة في مجلة Science Translational Medicine عام 2023 على نماذج حيوانية، لكن التجارب السريرية البشرية لا تزال في مراحلها الأولى.
أدوية L-Leucine: الحمض الأميني ليوسين (L-Leucine) أظهر في دراسات تجريبية قدرة على تعزيز ترجمة mRNA وتحسين إنتاج الكريات الحمراء عبر تفعيل مسار mTOR. تجارب سريرية صغيرة أُجريت وأظهرت بعض الاستجابة لدى عدد محدود من المرضى، لكن النتائج ليست حاسمة بعد.
مثبطات p53: نظراً لدور p53 المحوري في موت الخلايا السليفة الحمراء، يبحث العلماء في إمكانية استخدام مثبطات انتقائية لـ p53 لاستعادة تكوّن الكريات الحمراء. التحدي الأكبر هو أن تثبيط p53 قد يزيد خطر السرطان — لذلك يجب أن يكون انتقائياً ومضبوطاً بدقة.
تعديل الجينات بتقنية CRISPR: هناك اهتمام متزايد باستخدام تقنية كريسبر (CRISPR-Cas9) لتصحيح الطفرات الجينية مباشرة في الخلايا الجذعية للمريض. هذا النهج لا يزال في طور البحث المخبري (2024-2025)، لكنه يحمل وعداً هائلاً بتوفير علاج جذري أقل خطورة من زراعة النخاع.
اقرأ أيضاً:
- العلاج المناعي: كيف تبرمج جسمك ليدمر الخلايا السرطانية ذاتياً؟
- العلاج الموجه: ثورة الطب الحديث في قهر السرطان وتدمير الخلايا الخبيثة
ما بين الخرافة والحقيقة: معتقدات خاطئة عن متلازمة دياموند بلاكفان
❌ الخرافة: متلازمة دياموند بلاكفان تعني أن الطفل لن يعيش طويلاً.
✅ الحقيقة: كثير من المرضى يعيشون حياة طبيعية نسبياً مع العلاج المناسب. متوسط العمر المتوقع تحسّن على نحو ملحوظ بفضل التقدم في العلاج بالكورتيزون وزراعة النخاع وأدوية خلب الحديد. بعض المرضى يدخلون في هدأة تلقائية (Spontaneous Remission) بعد البلوغ.
❌ الخرافة: إعطاء الطفل مكملات الحديد سيعالج فقر الدم.
✅ الحقيقة: الحديد ليس المشكلة في هذا المرض — بل قد يكون جزءاً من المشكلة! معظم الأطفال المعتمدين على نقل الدم لديهم فائض حديد، وإعطاؤهم مكملات حديد إضافية قد يسرّع تلف الكبد والقلب. لا تعطِ طفلك أي مكمل حديد دون أمر صريح من الطبيب.
❌ الخرافة: المرض مُعدٍ أو ينتقل بالمخالطة.
✅ الحقيقة: متلازمة دياموند بلاكفان مرض وراثي جيني بحت. لا ينتقل بالعدوى أو المخالطة أو الهواء أو الطعام. هو طفرة في الحمض النووي للطفل، لا علاقة لها بأي عامل خارجي معدٍ.
❌ الخرافة: إذا لم يكن هناك تاريخ عائلي، فمستحيل أن يكون التشخيص صحيحاً.
✅ الحقيقة: كما ذكرنا، 40% إلى 45% من الحالات ناتجة عن طفرات تلقائية جديدة. غياب التاريخ العائلي لا ينفي التشخيص أبداً. المصدر: سجل دياموند بلاكفان الأميركي (DBAR).
❌ الخرافة: الأعشاب الطبيعية يمكن أن تعالج المتلازمة.
✅ الحقيقة: لا يوجد أي دليل علمي على أن أي عشبة أو مكمل عشبي يمكنه تحفيز نخاع العظم على إنتاج كريات حمراء في حالة خلل ريبوسومي جيني. بل إن بعض المكملات العشبية قد تتداخل مع الأدوية المستخدمة — كالكركم المركز الذي قد يعزز تأثير الكورتيزون على المعدة ويزيد خطر القرحة، أو الجنسنج الذي قد يؤثر في مستوى السكر لدى الأطفال على الكورتيزون. القاعدة الذهبية: لا تُدخل أي مكمل عشبي لنظام طفلك دون مراجعة طبيبه أولاً.
اقرأ أيضاً: الكركم (الذهب الأصفر): الفوائد العلاجية المثبتة علمياً وطرق استخدامه الصحيحة طبياً
هل تشير أعراض المتلازمة إلى أمراض أخرى في الجسم؟
فقر الدم الشديد عند الرضيع مع خلايا شبكية منخفضة ليس حكراً على متلازمة دياموند بلاكفان. هناك حالات أخرى يجب أن يستبعدها الطبيب قبل الوصول إلى التشخيص النهائي:
الأزمة اللاتنسجية العابرة (Transient Aplastic Crisis): تحدث غالباً بعد عدوى بفيروس بارفو B19 (Parvovirus B19) الذي يهاجم الخلايا السليفة الحمراء مؤقتاً. الفرق أنها عابرة وتتحسن تلقائياً خلال أسابيع، بينما فشل نخاع العظم عند الأطفال في متلازمة دياموند بلاكفان مزمن.
فقر الدم اللاتنسجي المكتسب (Acquired Aplastic Anemia): يؤثر في جميع خطوط إنتاج خلايا الدم (ليس الحمراء فقط)، وعادة يظهر في عمر أكبر. السبب غالباً مناعي ذاتي.
متلازمة بيرسون (Pearson Syndrome): اضطراب ميتوكوندري نادر يسبب فقر دم حرون مع خلل في البنكرياس. يتميز بوجود أرومات حديدية حلقية (Ring Sideroblasts) في النخاع.
أمراض المناعة الذاتية: بعض أمراض المناعة الذاتية عند الأطفال — خصوصاً الذئبة الحمامية الجهازية (Systemic Lupus Erythematosus – SLE) — قد تسبب فقر دم مع تثبيط النخاع. تُستبعد بفحوصات مناعية محددة.
الرسالة العملية: إذا شُخّص طفلك بـ “فقر دم غير معروف السبب”، لا تقبل بهذا التشخيص المبهم. اطلب تحويله إلى اختصاصي أمراض دم الأطفال لإجراء فحوصات تفريقية شاملة.
اقرأ أيضاً: انحلال الدم الوليدي: الأسباب، الأعراض، وخطوات العلاج والوقاية
ماذا عن التغذية في حالة متلازمة دياموند بلاكفان؟

⚠️ تحذير تغذوي: الطفل المصاب بهذه المتلازمة لديه احتياجات تغذوية خاصة تختلف عن الطفل السليم. لا تطبّق نصائح تغذية عامة دون استشارة اختصاصي تغذية سريرية مطّلع على حالة طفلك.
التغذية في هذا السياق تحمل تحديات فريدة. من جهة، يحتاج الطفل إلى تغذية كافية لدعم نموه المتأثر أصلاً بالمرض والعلاج. ومن جهة أخرى، يجب الحذر من الأطعمة الغنية بالحديد في حالة الأطفال الذين يعتمدون على نقل الدم؛ إذ إن إضافة حديد غذائي لجسم يعاني فائضاً فيه يشبه سكب الماء في وعاء يفيض.
نصائح تغذوية عملية للأطفال على نقل دم دوري:
- تجنب المكملات المحتوية على حديد: بما في ذلك الفيتامينات المتعددة المدعّمة بالحديد. اقرأ ملصق كل مكمل بعناية.
- لا حاجة لتقييد الحديد الغذائي الطبيعي بشكل صارم: تناول اللحوم والخضروات الورقية بكميات طبيعية مقبول. المشكلة في المكملات المركزة لا الطعام الطبيعي.
- التركيز على فيتامين C باعتدال: فيتامين C يزيد امتصاص الحديد من الأمعاء. تناول الفواكه الحمضية بكميات معتدلة مقبول، لكن تجنب مكملات فيتامين C بجرعات عالية مع الوجبات الغنية بالحديد.
- الكالسيوم وفيتامين D أساسيان: خصوصاً للأطفال على الكورتيزون، لحماية العظام من الهشاشة. استشر طبيبك عن الجرعة المناسبة لعمر طفلك.
- البروتين الكافي: ضروري لدعم النمو. مصادره: الحليب ومشتقاته، البيض، الدجاج، والبقوليات.
تنصح الدكتورة علا الأحمد — اختصاصية التغذية العلاجية في موقع وصفة طبية:
“الطفل المصاب بمتلازمة دياموند بلاكفان والمعتمد على نقل الدم يجب أن يُعامَل تغذوياً بمبدأ ‘التوازن الدقيق’: لا إفراط في الحديد ولا تفريط في البروتين والكالسيوم. أنصح الأهل بتحضير وجبات متنوعة غنية بالبروتين والخضروات الملونة، مع الحرص على إعطاء مكملات الكالسيوم وفيتامين D حسب توصية الطبيب — خصوصاً إذا كان الطفل يأخذ الكورتيزون.”
اقرأ أيضاً: غذاء طفلك في المدرسة: كيف تبني وجبة تعزز ذكاءه ومناعته وتجنبه التشتت
ما تكلفة علاج متلازمة دياموند بلاكفان تقريبياً؟
الحديث عن التكلفة المادية ليس ترفاً، بل حاجة واقعية للأسر التي تواجه مرضاً مزمناً. تتفاوت التكلفة على نحو كبير بحسب نوع العلاج والبلد ونظام التأمين الصحي:
العلاج بالكورتيزون: يُعَدُّ الأقل تكلفة نسبياً. شهرياً، قد لا يتجاوز 50 إلى 150 ريالاً سعودياً (15 إلى 40 دولاراً) لدواء البريدنيزولون وحده، لكن أضف إليه تكلفة المتابعة المخبرية الدورية وزيارات العيادة.
نقل الدم الدوري: كل جلسة نقل دم تكلف — في مستشفيات السعودية الحكومية — نحو 500 إلى 1,500 ريال. في المستشفيات الخاصة قد تصل إلى 3,000 ريال أو أكثر. ومع تكرار النقل كل 3 إلى 6 أسابيع، تتراكم التكلفة السنوية.
أدوية خلب الحديد: ديفيراسيروكس (Exjade) قد يكلف 3,000 إلى 8,000 ريال شهرياً بحسب الجرعة والوزن. في أميركا، السعر قد يتجاوز 3,000 دولار شهرياً دون تأمين.
زراعة الخلايا الجذعية: التكلفة الأعلى. في المراكز المتخصصة بالسعودية، قد تتراوح بين 300,000 إلى 700,000 ريال سعودي شاملة التحضير والرعاية بعد الزراعة. في أميركا وأوروبا، قد تصل إلى 200,000 إلى 500,000 دولار.
العوامل التي تتحكم في تفاوت السعر:
- نوع المتبرع (شقيق متطابق مقابل متبرع غير قريب).
- المضاعفات ومدة الإقامة في المستشفى.
- التأمين الصحي ومدى تغطيته.
- خبرة المركز الطبي وبنيته التحتية.
في السعودية، غالبية هذه التكاليف تُغطى في المستشفيات الحكومية والتخصصية للمواطنين السعوديين. أنصح الأهل بالتواصل مع مكتب الخدمات الاجتماعية في المستشفى لمعرفة الخيارات المتاحة.
التعايش مع المتلازمة: نصائح للأهل ومقدمي الرعاية
الدعم النفسي: ركيزة لا تقل أهمية عن الدواء
تشخيص طفلك بمرض وراثي نادر مثل متلازمة دياموند بلاكفان ليس خبراً سهلاً. الصدمة الأولى طبيعية، والشعور بالذنب — رغم أنه غير مبرر — شائع بين الأهل الذين يتساءلون: “هل فعلنا شيئاً خاطئاً؟” الإجابة العلمية واضحة: لا. الطفرة الجينية ليست نتيجة خطأ في التغذية أو السلوك أو البيئة. هي حدث جيني لا إرادة لأحد فيه.
اطلب دعماً نفسياً متخصصاً إذا شعرت بأن القلق يسيطر على حياتك اليومية. كثير من المراكز الطبية الكبرى في الرياض وجدة توفر خدمات الدعم النفسي لأسر مرضى الأمراض المزمنة. لا تتردد في طلب المساعدة — فأنت بحاجة إلى صحتك النفسية لتكون سنداً قوياً لطفلك.
الوقاية من العدوى: احتياطات يومية
الطفل على الكورتيزون أو في مرحلة ما بعد الزراعة يكون أكثر عرضة للعدوى. غسل اليدين المتكرر، تجنب الأماكن المزدحمة في مواسم الإنفلونزا، والحرص على التطعيمات الموصى بها (بعد مراجعة الطبيب، لأن بعض اللقاحات الحية قد تكون ممنوعة) — كلها خطوات بسيطة لكنها تحمي طفلك من مضاعفات خطيرة.
المتابعة الدورية مع فريق متعدد التخصصات
متلازمة دياموند بلاكفان ليست مرضاً يتابعه طبيب واحد. الطفل يحتاج إلى فريق يضم:
- اختصاصي أمراض دم الأطفال: لإدارة العلاج الأساسي.
- طبيب غدد صماء أطفال: لمراقبة النمو والبلوغ وتأثيرات تراكم الحديد على الغدد.
- طبيب قلب أطفال: لمتابعة وظائف القلب — خصوصاً مع نقل الدم المزمن أو عيوب القلب الخلقية.
- اختصاصي تغذية: لضبط النظام الغذائي.
- اختصاصي نفسي: لدعم الطفل والأسرة.
بروتوكول الفحص المبكر للأورام: خطوة استباقية لحماية طفلك
كما أشرنا سابقاً في التفاصيل الفسيولوجية، فإن مرضى متلازمة دياموند بلاكفان لديهم قابلية أعلى قليلاً للإصابة بأنواع معينة من سرطانات الدم (مثل متلازمة خلل التنسج النخاعي MDS) أو الأورام الصلبة (مثل ساركوما العظام وسرطان القولون) مقارنة بغيرهم.
هذه المعلومة ليست لإثارة الذعر، بل لبناء خطة مراقبة استباقية قوية. التوصيات الطبية الحديثة تلزم الأهل والفريق الطبي بالآتي:
- فحص سريري شامل مرة واحدة على الأقل سنوياً يبحث عن أي تضخم في الغدد الليمفاوية، أو آلام عظمية غير مبررة، أو تغيرات في تعداد الدم (مثل انخفاض مفاجئ في الصفائح الدموية أو الكريات البيضاء والذي لا يميز المرض عادةً).
- خزعة نخاع عظم دورية: يُنصح بإجرائها فوراً في حال حدوث أي تدهور مفاجئ في استجابة المريض للعلاج، أو ظهور خلايا غير طبيعية في تحليل الدم الروتيني.
- التوعية المبكرة للبالغين: عند وصول المريض لمرحلة البلوغ، يجب توجيهه للبدء في فحوصات الكشف المبكر عن سرطان القولون في سن مبكرة (أبكر من التوصيات العامة للسكان).
هل تعلم؟ بعض المرضى — بنسبة تتراوح بين 15% و25% — يدخلون في هدأة تلقائية (Spontaneous Remission) عادة بعد سن البلوغ، فيتوقف اعتمادهم على الكورتيزون أو نقل الدم تماماً. الأسباب غير مفهومة بالكامل، لكنها تمنح الأمل. المصدر: سجل دياموند بلاكفان (DBAR).
الخطة العملية للتعامل مع متلازمة دياموند بلاكفان: تعليمات قبل مغادرة العيادة
- احتفظ بملف طبي منظم: يشمل كل نتائج التحاليل، مواعيد نقل الدم، جرعات الأدوية، ومنحنيات النمو. حمّل تطبيقاً صحياً على هاتفك لتسجيل هذه البيانات.
- لا تفوّت موعد الفيريتين: إذا كان طفلك على نقل دم دوري، تأكد من فحص مستوى الفيريتين كل 3 أشهر على الأقل. ارتفاعه فوق 1,000 نانوغرام/مل يستوجب تفعيل أو تكثيف العلاج الخالب.
- راقب علامات الخطر: ارتفاع حرارة فجائي، شحوب مفاجئ شديد، ضيق تنفس، أو نزف غير عادي — كلها تستوجب التوجه إلى الطوارئ فوراً.
- تابع فحوصات السمع والبصر سنوياً: خصوصاً إذا كان طفلك يأخذ أدوية خلب الحديد.
- تأكد من تطعيمات طفلك: بعد مراجعة الطبيب لتحديد أيها آمن في حالته.
- تواصل مع مجموعات الدعم: هناك مجموعات عبر الإنترنت لأسر مرضى متلازمة دياموند بلاكفان، مثل Diamond Blackfan Anemia Foundation، توفر دعماً معنوياً ومعلومات محدثة.
- خطط للحياة المدرسية: تحدث مع إدارة مدرسة طفلك عن حالته، واشرح لهم ما قد يحتاجه من تسهيلات — كالإعفاء من الأنشطة البدنية الشاقة في أيام نقل الدم.
اقرأ أيضاً:
- صندوق الإسعافات الأولية: كيف تنقذ حياة من تحب في اللحظات الحرجة؟
- الإسعافات الأولية: خطوات وإجراءات طبية تنقذ حياتك وقت الطوارئ
الوصفة الطبية من موقعنا
- حماية العظام من الداخل: الكورتيزون يسرق الكالسيوم من عظام طفلك ببطء. تأكد من حصوله على 500 إلى 1,000 ملغ كالسيوم يومياً (حسب عمره) مع 400 إلى 1,000 وحدة دولية من فيتامين D3. هذا المزيج يدعم عمل بانيات العظم (Osteoblasts) ويقلل تأثير الكورتيزون التدميري على الكثافة العظمية.
- النوم العميق ليس ترفاً: هرمون النمو (Growth Hormone) يُفرَز على نحو رئيس في أثناء النوم العميق. الطفل المصاب بقصر قامة مرتبط بالمتلازمة يحتاج إلى 10 إلى 12 ساعة نوم ليلي منتظم في غرفة مظلمة وهادئة. أطفئ الشاشات قبل النوم بساعة على الأقل لتعزيز إفراز الميلاتونين (Melatonin) الذي ينظّم الإيقاع اليوماوي (Circadian Rhythm).
- حركة خفيفة ومنتظمة: ليس المطلوب تمريناً رياضياً عنيفاً. المشي 20 إلى 30 دقيقة يومياً أو اللعب الحر يعزز الدورة الدموية، ويحفز إفراز عامل النمو الشبيه بالإنسولين (IGF-1) الذي يدعم النمو الطولي، ويحسّن المزاج بتحفيز الإندورفينات.
- تقليل الحمل الالتهابي الغذائي: تجنب الأطعمة فائقة المعالجة (Ultra-Processed Foods) والسكريات المضافة المفرطة. هذه الأطعمة ترفع مستويات السيتوكينات الالتهابية (Inflammatory Cytokines) مثل IL-6 وTNF-α، ما يزيد العبء على جسم يكافح أصلاً خللاً مزمناً. ركّز على الخضروات الملونة والفواكه الطازجة والحبوب الكاملة.
- الدعم النفسي المبكر بمنهجية: لا تنتظر حتى يظهر على طفلك اكتئاب أو انسحاب اجتماعي. ابدأ بجلسات لعب علاجي (Play Therapy) أو حكايات مصوّرة عن أطفال أبطال يتعايشون مع أمراض مزمنة. التحفيز العاطفي الإيجابي المبكر يقوي شبكات الدماغ المرتبطة بالمرونة النفسية (Psychological Resilience).
- مراقبة الوزن بذكاء: الكورتيزون يزيد الشهية ويعيد توزيع الدهون. بدلاً من تقييد الطعام — وهو ضار لطفل ينمو — ركّز على نوعية الطعام: بروتين عالي الجودة، ودهون صحية (زيت زيتون، أفوكادو)، وتقليل الوجبات الخفيفة السكرية. الهدف ليس الحرمان بل الحماية الأيضية.
اقرأ أيضاً: حاسبة النوم حسب العمر
ما الذي يجب أن يعرفه الأهل في السعودية تحديداً؟
المملكة العربية السعودية تتميز بانتشار نسبي أعلى للأمراض الوراثية بسبب ارتفاع معدلات زواج الأقارب. وعلى الرغم من أن متلازمة دياموند بلاكفان تنتقل بالوراثة السائدة في الغالب (وليس المتنحية التي تزيد مع زواج الأقارب)، إلا أن وعي الأسر بأهمية الفحص الجيني قبل الزواج والفحص الوراثي عند الإنجاب يبقى أمراً محورياً.
في عام 2024، أطلقت المملكة مبادرات ضمن رؤية 2030 لتعزيز الفحص الجيني الشامل للأمراض النادرة، بما في ذلك توسيع نطاق فحص حديثي الولادة. إذا كان في عائلتك تاريخ لأي مرض وراثي دموي، فاستشر طبيب وراثة قبل التخطيط للحمل.
ومضة سعودية: يُوفّر مستشفى الملك فيصل التخصصي ومركز الأبحاث بالرياض عيادة متخصصة لأمراض فشل نخاع العظم الوراثية، تضم فريقاً متكاملاً من أطباء الدم والوراثة والتغذية والدعم النفسي. استفسر عن الإحالة من طبيب طفلك.
اقرأ أيضاً: تأخر الكلام واللغة عند الأطفال: الأسباب الطبية والعلامات التحذيرية وأحدث طرق العلاج
يقول الدكتورة سوزان عبد الحميد السعدي — استشارية أمراض الدم وزرع الخلايا الجذعية في موقع وصفة طبية:
“متلازمة دياموند بلاكفان مرض نادر لكنه ليس بلا أمل. الأطفال الذين يُشخَّصون مبكراً ويُتابَعون بانتظام مع فريق متعدد التخصصات يحققون نتائج ممتازة. أشجع كل أسرة على بناء علاقة ثقة طويلة الأمد مع فريقها الطبي، وعدم التردد في طرح أي سؤال مهما بدا بسيطاً. طفلكم يستحق أفضل رعاية — وأنتم الركيزة الأولى لنجاحها.”
الخاتمة: رسالة أمل مبنية على العلم
إذا وصلت إلى هنا، فأنت أب أو أم أو مقدم رعاية يستثمر وقته في فهم حالة نادرة ومعقدة — وهذا في حد ذاته أول خطوة نحو رعاية أفضل لطفلك. متلازمة دياموند بلاكفان مرض مزمن، نعم. لكنه مرض يملك الطب أدوات حقيقية لمواجهته: من الكورتيزون إلى نقل الدم، ومن أدوية خلب الحديد إلى زراعة النخاع، ومن الأبحاث الجينية الواعدة إلى تقنيات كريسبر المستقبلية.
التزامك بخطة العلاج، ومتابعتك الدورية مع الفريق الطبي، ودعمك النفسي لطفلك ولنفسك — هذه الثلاثية هي مفتاح جودة الحياة. لا تدع التشخيص يحدد مستقبل طفلك؛ بل دعه يكون نقطة البداية لفهم أعمق ورعاية أذكى.
هل حدَّثت طبيب طفلك اليوم عن آخر نتيجة فيريتين، وهل ناقشت معه خيار الفحص الجيني للعائلة؟
نعم، في حالة زراعة نخاع العظم من شقيق متطابق، تتجاوز نسبة الشفاء التام 90% عند الأطفال دون سن العاشرة. كما يدخل 15% إلى 25% من المرضى في هدأة تلقائية بعد البلوغ دون علاج.
يحتاج معظم الأطفال الذين لا يستجيبون للكورتيزون إلى نقل دم كل 3 إلى 6 أسابيع للحفاظ على مستوى هيموغلوبين لا يقل عن 8 غ/دل. يحدد الطبيب التكرار بناءً على معدل انخفاض الهيموغلوبين لدى كل طفل.
نعم، يرتفع خطر الإصابة بمتلازمة خلل التنسج النخاعي (MDS) وابيضاض الدم النقوي الحاد (AML) بنحو 5 أضعاف مقارنة بالسكان الطبيعيين، نتيجة الخلل المزمن في مسار p53. المتابعة المنتظمة مع الطبيب ضرورية للكشف المبكر.
إذا كانت الطفرة الجينية معروفة في أحد الوالدين، يمكن إجراء تشخيص قبل الولادة عبر اختبار الزغابات المشيمية (CVS) أو بزل السلى (Amniocentesis). أما بدون تاريخ عائلي معروف، فلا يوجد حالياً فحص روتيني قبل الولادة.
نعم، لكن يحمل كل طفل خطر 50% لوراثة الطفرة إذا كانت الوراثة سائدة. يُنصح بمشورة وراثية متخصصة قبل التخطيط للحمل، وقد يُقترح التشخيص الجيني ما قبل الزرع (PGT) للأزواج الراغبين في تقليل هذا الخطر.
في متلازمة دياموند بلاكفان يتأثر خط الكريات الحمراء فقط مع بقاء الكريات البيضاء والصفائح طبيعية. في فقر الدم اللاتنسجي يتأثر نخاع العظم بأكمله ويتوقف عن إنتاج جميع خلايا الدم. السبب والعمر والعلاج مختلفة جوهرياً.
نعم، الاستخدام المطوّل للكورتيزون بجرعات عالية يُبطئ النمو الطولي. لهذا يسعى الأطباء لأقل جرعة فعالة، ويُنصح بمراقبة منحنى النمو شهرياً وتعويض الكالسيوم وفيتامين D بانتظام.
معظم التطعيمات المعطّلة (غير الحية) آمنة. لكن اللقاحات الحية المخففة — كالحصبة والنكاف والحماق — قد تكون ممنوعة عند الأطفال على جرعات كورتيزون عالية أو بعد زراعة النخاع. استشر طبيب طفلك قبل أي تطعيم.
توجد مجموعات دعم عربية على منصات التواصل الاجتماعي يمكن إيجادها بالبحث عن “متلازمة دياموند بلاكفان” على فيسبوك وواتساب. دولياً، تُعَدُّ Diamond Blackfan Anemia Foundation (dbafoundation.org) الأكثر شمولاً وتقدم دعماً متعدد اللغات.
يعتمد ذلك على مستوى الهيموغلوبين وحالة الطفل. في أيام قرب موعد نقل الدم حين ينخفض الهيموغلوبين، يُنصح بتقليل النشاط الشاق. بعد نقل الدم، يمكن ممارسة أنشطة خفيفة إلى معتدلة بموافقة الطبيب وتبليغ إدارة المدرسة.
يلتزم موقع وصفة طبية بأعلى معايير الدقة العلمية والشفافية في تقديم المحتوى الطبي. إليك كيف نضمن ذلك:
- يُكتب كل مقال بالاستناد إلى مصادر علمية محكّمة من مجلات طبية دولية معتمدة (PubMed، Cochrane، NEJM، Lancet، Blood وغيرها).
- تُراجَع المقالات الطبية من قِبل أطباء متخصصين ومعتمدين قبل نشرها، ويُشار إلى أسمائهم في كل مقال.
- يخضع كل مقال لتدقيق علمي وتدقيق مصادر ومراجعة لغوية قبل النشر.
- نحرص على تحديث المقالات بصفة دورية لمواكبة أحدث المستجدات الطبية.
- نُفصح بوضوح عن أي تعارض محتمل في المصالح ونلتزم بعدم الترويج لأي منتج دوائي أو تجاري بشكل مموّه.
- نتبع معايير HONcode وإرشادات المحتوى الصحي الرقمي الصادرة عن منظمة الصحة العالمية (WHO).
- Diamond Blackfan Anemia Registry (DBAR) 2024 — الإرشادات التشخيصية والعلاجية لمتلازمة دياموند بلاكفان الصادرة عن السجل الأميركي الرسمي للمرض.
- National Institutes of Health — NIH (USA) 2024 — بروتوكولات إدارة تراكم الحديد الناتج عن نقل الدم المزمن في أمراض فشل نخاع العظم الوراثية.
- European Hematology Association (EHA) 2023 — دليل ممارسة سريرية لعلاج الاضطرابات الريبوسومية الوراثية وخلايا فشل النخاع عند الأطفال.
- U.S. Food and Drug Administration (FDA) 2022 — النشرة الدوائية الرسمية المعتمدة لأدوية خلب الحديد: ديفيراسيروكس (Jadenu / Exjade) وديفيريبرون (Ferriprox).
- وزارة الصحة السعودية — المديرية العامة للأمراض النادرة 2024 — الإطار الوطني لرعاية الأمراض الوراثية النادرة ضمن مبادرات رؤية 2030 للفحص الجيني الشامل لحديثي الولادة.
- World Health Organization (WHO) — Genomics and Rare Diseases 2023 — إطار منظمة الصحة العالمية لدعم تشخيص وعلاج الأمراض الوراثية والنادرة على المستوى العالمي.
المصادر والمراجع
الدراسات والأوراق البحثية
- Da Costa, L., Narla, A., & Bhatt, A. P. (2018). Diamond-Blackfan Anemia. Blood, 131(18), 2028–2037. DOI: 10.1182/blood-2017-12-822437
— مراجعة شاملة للآليات الجزيئية والسريرية لمتلازمة دياموند بلاكفان. - Vlachos, A., & Muir, E. (2010). How I treat Diamond-Blackfan Anemia. Blood, 116(19), 3715–3723. DOI: 10.1182/blood-2010-02-251090
— بروتوكول علاجي عملي من أبرز خبراء المرض عالمياً. - Ulirsch, J. C., et al. (2018). The genetic landscape of Diamond-Blackfan Anemia. American Journal of Human Genetics, 103(6), 930–947. DOI: 10.1016/j.ajhg.2018.10.027
— دراسة جينومية واسعة تحدد الطفرات المسببة للمرض. - Bartels, M., et al. (2021). Outcomes of hematopoietic stem cell transplantation in Diamond-Blackfan Anemia. Blood, 138(Supplement 1), 342. DOI: 10.1182/blood-2021-148235
— نتائج زراعة النخاع عند أطفال المتلازمة ونسب النجاح. - Khajuria, R. K., et al. (2018). Ribosome levels selectively regulate translation and lineage commitment in human hematopoiesis. Cell, 173(1), 90–103. DOI: 10.1016/j.cell.2018.02.036
— دراسة رائدة توضح لماذا يتأثر خط الكريات الحمراء تحديداً بالخلل الريبوسومي. - Aspesi, A., & Ellis, S. R. (2019). Rare ribosomopathies: insights into mechanisms of cancer. Nature Reviews Cancer, 19(4), 228–238. DOI: 10.1038/s41568-019-0105-0
— مراجعة العلاقة بين أمراض الريبوسومات وخطر السرطان.
الجهات الرسمية والمنظمات
- National Institutes of Health (NIH). (2024). Diamond-Blackfan Anemia – Genetic and Rare Diseases Information Center.
— صفحة مرجعية شاملة عن المرض من المعاهد الوطنية للصحة الأميركية. - Diamond Blackfan Anemia Registry (DBAR). (2024). About DBA.
— السجل الأميركي الرسمي للمرض وأكبر قاعدة بيانات سريرية عالمية. - U.S. Food and Drug Administration (FDA). (2022). Deferasirox (Jadenu) Prescribing Information.
— النشرة الدوائية الرسمية لدواء ديفيراسيروكس. - Orphanet. (2024). Diamond-Blackfan Anemia.
— قاعدة بيانات أوروبية للأمراض النادرة. - World Health Organization (WHO). (2023). Genomics and Rare Diseases.
— إطار منظمة الصحة العالمية لدعم الأمراض الوراثية والنادرة.
الكتب والموسوعات العلمية
- Nathan, D. G., Orkin, S. H., et al. (2015). Nathan and Oski’s Hematology and Oncology of Infancy and Childhood (8th ed.). Elsevier.
— المرجع الأشمل في أمراض دم الأطفال، يتضمن فصلاً مفصلاً عن متلازمة دياموند بلاكفان. - Lipton, J. M., & Ellis, S. R. (2010). Diamond-Blackfan Anemia. In Inherited Bone Marrow Failure Syndromes (pp. 45–93). Springer.
— فصل أكاديمي متعمق ضمن كتاب متخصص في متلازمات فشل النخاع الموروثة. - Hoffman, R., et al. (2018). Hematology: Basic Principles and Practice (7th ed.). Elsevier.
— مرجع أكاديمي معتمد في أمراض الدم يغطي الآليات الجزيئية والسريرية.
مقالات علمية مبسطة
- Narla, A., & Ebert, B. L. (2018). Translational medicine: ribosomopathies. Blood, 131(18), 1953–1957. DOI: 10.1182/blood-2017-11-742411
— مقالة مبسطة نسبياً تشرح مفهوم أمراض الريبوسومات لجمهور طبي واسع.
قراءات إضافية ومصادر للتوسع
- Clinton, C., & Gazda, H. T. (2009, updated 2023). Diamond-Blackfan Anemia. In GeneReviews®. University of Washington, Seattle.
— رابط GeneReviews
— لماذا نقترح عليك قراءته؟ هذا المرجع يُحدَّث دورياً ويقدم أعمق مراجعة جينية وسريرية متاحة مجاناً عبر الإنترنت، ويُعَدُّ المرجع الأول لأطباء الوراثة في العالم. - Lipton, J. M., & Ellis, S. R. (2009). Diamond-Blackfan Anemia: Diagnosis, Treatment, and Molecular Pathogenesis. Hematology/Oncology Clinics of North America, 23(2), 261–282.
— DOI: 10.1016/j.hoc.2009.01.004
— لماذا نقترح عليك قراءته؟ ورقة مراجعة شاملة تربط بين المسار الجزيئي والتطبيق السريري، مثالية لطلاب الطب والباحثين الذين يريدون فهم “الصورة الكبيرة”. - Danilova, N., & Gazda, H. T. (2015). Ribosomopathies: how a common root can cause a tree of pathologies. Disease Models & Mechanisms, 8(9), 1013–1026.
— DOI: 10.1242/dmm.020529
— لماذا نقترح عليك قراءته؟ يمنح القارئ منظوراً أوسع عن عائلة “أمراض الريبوسومات” ككل، ويوضح لماذا طفرة واحدة في بروتين ريبوسومي يمكن أن تسبب طيفاً واسعاً من الأمراض — ليس فقط متلازمة دياموند بلاكفان.
إذا وجدت هذا المقال مفيداً، شاركه مع أسرة أخرى قد تحتاجه. كل مشاركة قد تساعد أباً أو أماً يبحث في الإنترنت عن إجابات لم يجدها بعد. ولا تنسَ أن الاشتراك في نشرة موقع وصفة طبية يبقيك على اطلاع بأحدث المستجدات الطبية في أمراض الدم والأمراض الوراثية النادرة عند الأطفال.
المحتوى المنشور في موقع وصفة طبية هو للأغراض التثقيفية والمعلوماتية العامة فقط، ولا يُعَدُّ بديلاً عن التشخيص الطبي أو النصيحة العلاجية أو الوصفة الطبية المقدمة من طبيب مرخص. جميع المعلومات مبنية على مصادر علمية محكّمة وآراء أطباء متخصصين، غير أنها لا تأخذ في الاعتبار الحالة الفردية لكل مريض.
لا تتخذ أي قرار علاجي — بما في ذلك تناول الأدوية أو تعديل جرعاتها أو إيقافها — بناءً على ما تقرأه هنا دون الرجوع إلى طبيبك أو مقدم الرعاية الصحية المختص. في حالات الطوارئ الطبية، اتصل بالإسعاف أو توجه فوراً إلى أقرب مرفق صحي.
يتحمل القارئ مسؤولية كاملة عن أي قرار يتخذه اعتماداً على المحتوى دون استشارة طبية مسبقة. موقع وصفة طبية وفريقه التحريري غير مسؤولين عن أي نتائج مباشرة أو غير مباشرة تنجم عن ذلك.